溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這篇文章主要介紹“如何使用python Genome Tracks可視化hi-c數據”,在日常操作中,相信很多人在如何使用python Genome Tracks可視化hi-c數據問題上存在疑惑,小編查閱了各式資料,整理出簡單好用的操作方法,希望對大家解答”如何使用python Genome Tracks可視化hi-c數據”的疑惑有所幫助!接下來,請跟著小編一起來學習吧!
可視化是數據分析中非常重要的一個環節,對于NGS分析數據的可視化,最常用的就是各種基因組瀏覽器了,既有UCSC, GBrowse等基于web的基因組瀏覽器,也有igvtools等本地化的圖形界面軟件。對于Hi-C數據,在前面的文章中也介紹過基于web的WashU Epigenome Browser基因組瀏覽器和本地化的juicebox軟件。
熟練掌握其中一個軟件的用法就可以滿足大部分的需求了,但是作為一個生信分析的極客,總感覺還是需要一款命令行工具來提高效率。python和R都擁有非常強大的可視化能力,今天介紹一款基于python語言的軟件pyGenomeTracks, 一款原汁原味的命令行工具,擁有和基因組瀏覽器相同的展現形式,網址如下
https://github.com/deeptools/pyGenomeTracks
該軟件支持可視化以下幾種信息
bigwig
bed
bedgraph
links
Hi-C matrices
采用該軟件可視化的效果圖如下
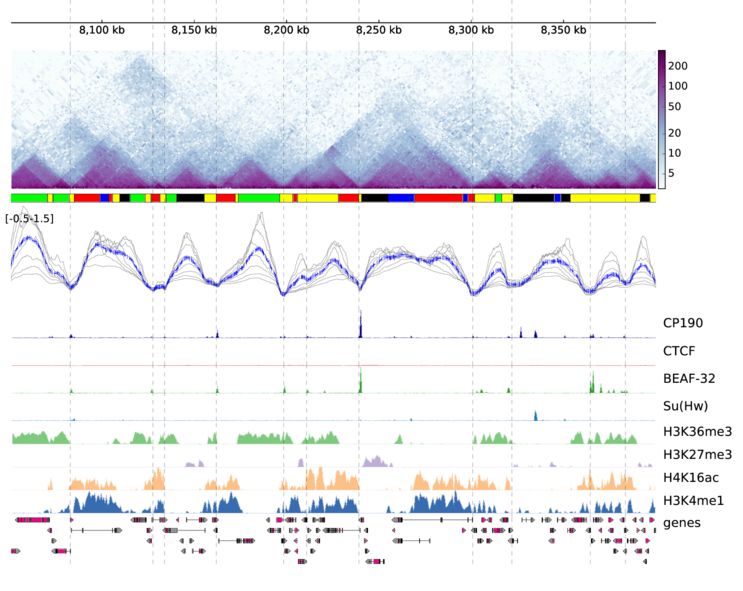
和基因組瀏覽器一樣的展現形式,每一層為一個track。該軟件采用配置文件的形式來配置需要展示的文件信息,每個需要展示的文件和對應的參數都寫在一個標簽下,具體寫法如下
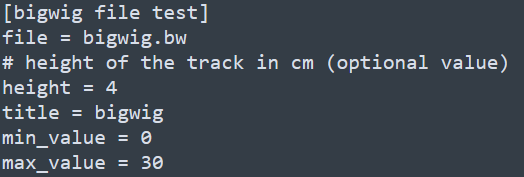
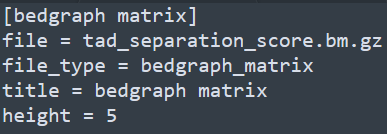
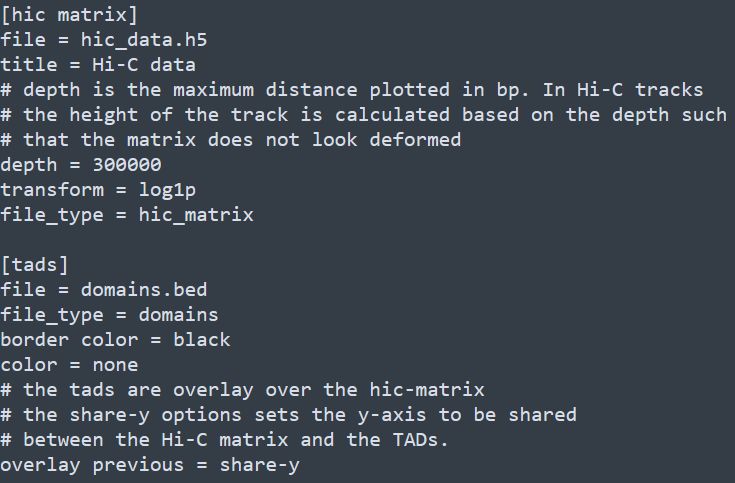
除此之后,還有x-axis和spacer等標簽,分別對應x軸和兩個tracks之間的空格區域。下方如下
[spacer]
[x-axis]
where = top編輯好配置文件之后,就可以運行了,用法如下
pyGenomeTracks \
--tracks tracks.ini \
--region chr2:10,000,000-11,000,000 \
--outFileName output.pdftracks參數指定配置文件的名稱,region參數指定需要可視化的基因組區域,outFileName參數指定輸出文件的名稱。為了達到美觀的效果,有許多的參數需要調整,更多細節請參考官方文檔和示例。
一個hi-c數據可視化的效果圖如下
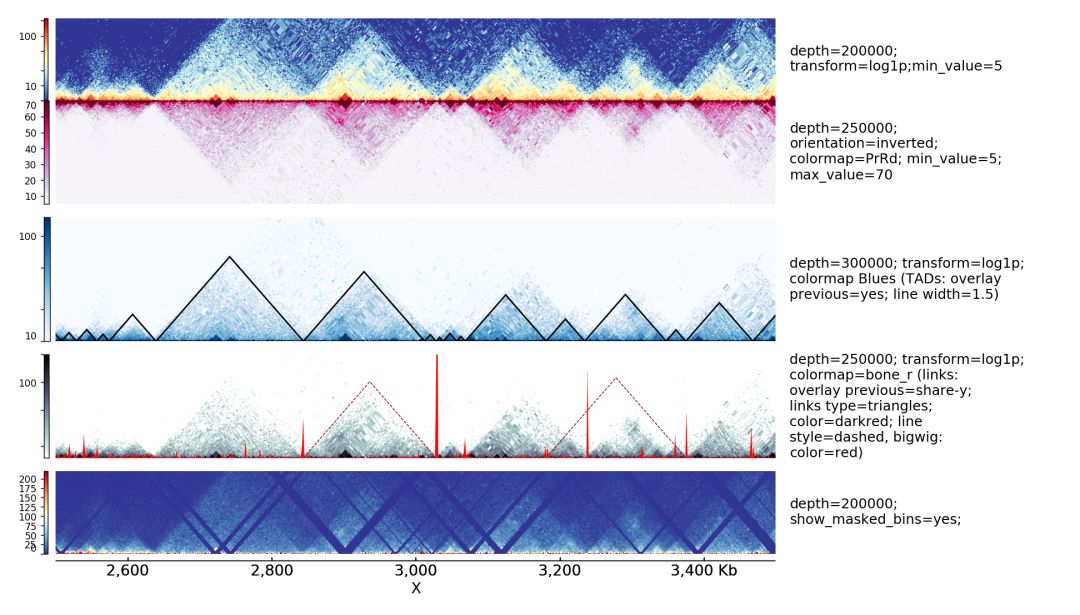
通過該軟件,可以高效的展示hi-c數據。
到此,關于“如何使用python Genome Tracks可視化hi-c數據”的學習就結束了,希望能夠解決大家的疑惑。理論與實踐的搭配能更好的幫助大家學習,快去試試吧!若想繼續學習更多相關知識,請繼續關注億速云網站,小編會繼續努力為大家帶來更多實用的文章!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





