溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這篇文章主要講解了“ALLPATHS-LG怎么安裝使用”,文中的講解內容簡單清晰,易于學習與理解,下面請大家跟著小編的思路慢慢深入,一起來研究和學習“ALLPATHS-LG怎么安裝使用”吧!
!
ALLPATHS-LG 是由Broad Institiute研究所發明的一款基因組組裝軟件,不論是細菌/真菌等小型基因組,還是動植物等大型基因組的組裝,它都能夠勝任。
和其他組裝軟件不同的是,allpaths-lg要求至少兩個文庫
第一個文庫的插入片段長度不能超過測序讀長的兩倍,這樣可以保證雙端測序的reads之間存在overlap,這樣的文庫類型稱之為fragment
第二個文庫的插入片段通常大于3kb,超長讀長有利于基因組的組裝,這樣的文庫類型稱之為jumping
除了插入片段外,allpaths-lg對測序深度也有要求,推薦100X以上。
在組裝時,對于硬件資源也有一定的要求,對于哺乳動物基因組,建議內存大小為512G, 對于小基因組,建議內存大小32G。
安裝過程如下
wget ftp://ftp.broadinstitute.org/pub/crd/ALLPATHS/Release-LG/latest_source_code/allpathslg-52488.tar.gz tar xzvf allpathslg-52488.tar.gz cd allpathslg-52488/ ./configure --prefix=$(pwd) make make install
安裝好之后,在bin目錄下可以找到程序的可執行文件。為了方便調用,可以把bin目錄添加到PATH環境變量中。官方提供了小的測試數據集, 可以幫助我們了解軟件的用法
wget ftp://ftp.broadinstitute.org/pub/crd/ALLPATHS/Release-LG/test.genome.tar.gz
allpaths-lg的運行分成以下兩步
在bin目錄下,有一個名為PrepareAllPathsInputs.pl的可執行文件,這個文件就是用來準備輸入文件的。這個文件需要讀取以下兩個文件
in_groups.csv,示例如下
file_name, library_name, group_name seq/frags.?.fastq, Solexa-25396, frags seq/jumps.?.fastq, Solexa-11542, jumps
逗號分隔的3列文件,group_name代表每組的唯一的ID,library_name代表文庫的名字, file_name 代表序列文件,對于雙端測序的文件,可以使用通配符來表示R1端和R2端。
in_libs.csv, 示例如下
library_name, project_name, organism_name, type, paired, frag_size, frag_stddev, insert_size, insert_stddev, read_orientation, genomic_start, genomic_end Solexa-25396, test, test.genome, fragment, 1, 180, 10, , , inward, 0, 0 Solexa-11542, test, test.genome, jumping, 1, , , 3000, 500, outward, 0, 0
逗號分隔的12列文件,library_name和in_groups.csv文件中的文庫名字相同,project_name代表項目名稱,organism_name代表組裝的物種名稱,type代表文庫類型,fragment代表插入片段短,存在overlap的文庫;jumping代表插入片段非常長的文庫,paired代表測序類型,0表示單端測序,1表示雙端測序;frag_size和frag_stddev只針對fragment文庫,分別表示插入片段長度的平均數和方差;insert_size和insert_stddev只針對jumping文庫,分別代表jumping文庫插入片段長度的均值和方差;read_orientation代表測序方向,genomic_start和genome_end用來過濾序列,小于genome_start和大于genome_end的序列會被過濾掉,在實際使用時,直接填0就可以了。
對于fragment和jumping 文庫,測序方向分別對應inward和outward。
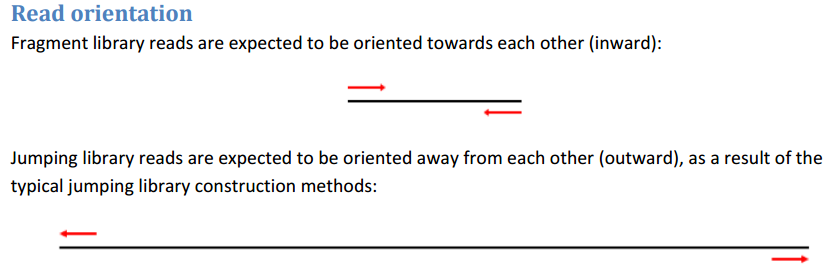
以上兩個文件根據自己的數據填寫好之后,就可以運行下面的代碼了
PrepareAllPathsInputs.pl \ DATA_DIR=$PWD/test.genome/data \ PLOIDY=1 \ IN_GROUPS_CSV=in_groups.csv \ IN_LIBS_CSV=in_libs.csv \ GENOME_SIZE=200000 \ OVERWRITE=True
DATA_DIR代表數據的存放目錄,要去必須是絕對路徑,test.genome必須和csv文件中的organism_name相同。PLOIDY代表染色體倍性,allpaths-lg目前只支持單倍體和二倍體。運行結束后,在輸出目錄會生成如下文件
├── frag_reads_orig.fastb ├── frag_reads_orig.pairs ├── frag_reads_orig.qualb ├── frag_reads_orig.source.txt ├── jump_reads_orig.fastb ├── jump_reads_orig.pairs ├── jump_reads_orig.qualb ├── jump_reads_orig.source.txt ├── ploidy └── read_cache
每個文庫的序列會生成對應的.fastb, .pairs, qualb三個文件;ploidy 記錄染色體倍性;read_cache 是臨時目錄。
準備好輸入文件之后,就可以進行組裝了,命令如下
RunAllPathsLG \ PRE=$PWD\ REFERENCE_NAME=test.genome\ DATA_SUBDIR=data\ RUN=run\ SUBDIR=test\ TARGETS=standard\ OVERWRITE=True
上述命令中的5個參數構成了如下的目錄結構
PRE/REFERENCE_NAME/DATA_SUBDIR/RUN/SUBDIR
allpaths-lg通過這樣的目錄結構來存放多個基因組組裝的結果。
組裝的結果保存在SUBDIR 目錄下,final.contigs.fasta對應contig的結果,final.assembly.fasta對應scaffold的結果。
感謝各位的閱讀,以上就是“ALLPATHS-LG怎么安裝使用”的內容了,經過本文的學習后,相信大家對ALLPATHS-LG怎么安裝使用這一問題有了更深刻的體會,具體使用情況還需要大家實踐驗證。這里是億速云,小編將為大家推送更多相關知識點的文章,歡迎關注!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





