溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
本篇文章為大家展示了如何使用CIRI識別環狀RNA,內容簡明扼要并且容易理解,絕對能使你眼前一亮,通過這篇文章的詳細介紹希望你能有所收獲。
在最初的環狀RNA研究中,認為環狀RNA都是由exon通過反向剪切構成的,稱之為exonic circRNA,只有這樣的環狀RNA能夠由PCR反應驗證出來的。
CIRI是一款環狀RNA檢測軟件,通過該軟件的預測結果,學者第一次用實驗驗證出了intronic circRNA和intergenic circRNA。該軟件操作簡便,準確度高,是非常流行的一款環狀RNA檢測軟件。
該軟件至少需要兩個輸入文件,基因組的fasta序列和測序數據比對產生的sam文件,需要注意的是,輸入的sam文件必須是由bwa-mem算法比對產生的 。分析的pipeline示意如下
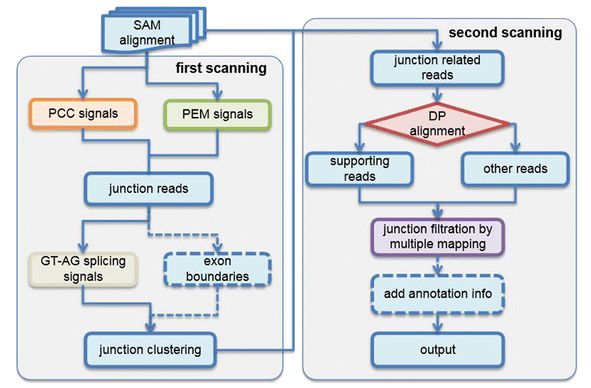
對于輸入的sam文件,需要經過兩次掃描,在第一次掃描時,根據雙端數據的比對情況篩選候選的環狀RNA,這一步通過判斷SAM文件中CIGAR那一列的值來實現,本質上是檢測覆蓋環狀RNA連接點處的junction reads,根據測序讀長和連接點處包含的基因組區域的特征,分成以下3種模型
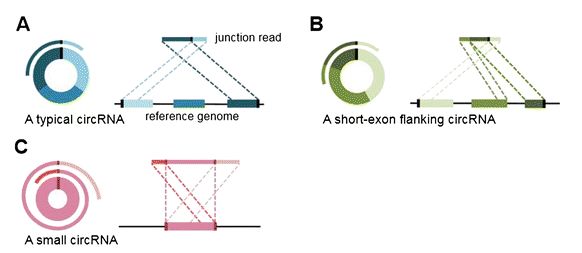
圖A表示junction read只覆蓋了起始外顯子和終止外顯子的部分序列,這兩部分reads在基因組上的比對位置是相反的,絕大部分的環狀RNA都符合這種模型。
圖B表示junction read除了覆蓋了起始外顯子和終止外顯子的兩部分序列外,還覆蓋了中間的一個外顯子的部分序列,這種情況下reads可以分成3個部分比對到基因組上。
圖C表示junction read除了覆蓋了整個環狀RNA外,還重復又讀了一部分序列,這個只有當環狀RNA的序列長度小于測序讀長時才可能出現。
該軟件將以上3種模型定義為paired chiastic clipping signals,簡稱PCC信號,如果一條reads比對情況符合以上任意一種,就認為該reads是一條環狀RNA的junction reads。
為了提高準確性,識別到junciton reads之后,還會結合雙端序列比對的質量paired end mapping即PEM和GT-AG保守的剪切位點進行過濾,示意圖如下
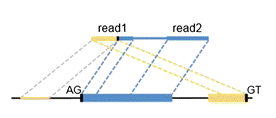
只保留比對質量較高,且頭尾符合AG-GT剪切信號的junciton reads進入下游分析,在第二次掃描SAM文件的過程中,通過動態規劃算法給出最終的環狀RNA預測結果,如果提供了GTF文件,還會對環狀RNA進行注釋。
該軟件的使用步驟如下
代碼如下
bwa mem \ -T 19 \ -t 5 hg19_index \ R1.fastq.gz R2.fastq.gz \ > align.sam
CIRI2.pl \ -T 20 \ -F hg19.fa \ -A hg19.gtf \ -I align.sam \ -O circRNA.xls
輸出結果如下所示
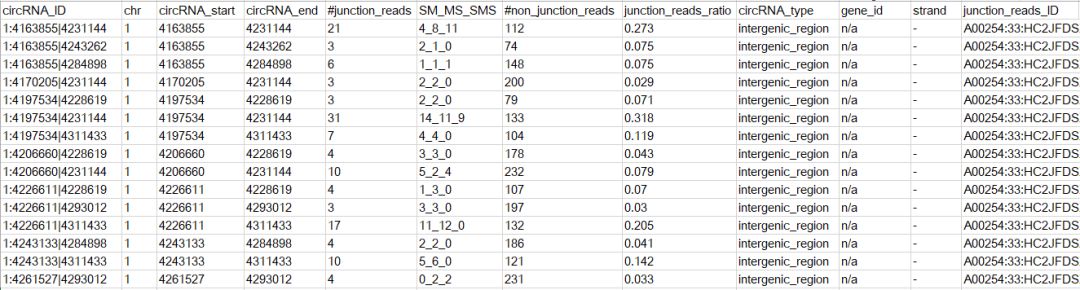
在后續驗證時,可以挑選表達量較高的來驗證,在軟件對應的文章中,挑選了junction reads數大于5的環狀RNA來進行驗證。
上述內容就是如何使用CIRI識別環狀RNA,你們學到知識或技能了嗎?如果還想學到更多技能或者豐富自己的知識儲備,歡迎關注億速云行業資訊頻道。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





