溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
circRNA_finder中如何識別環狀RNA,相信很多沒有經驗的人對此束手無策,為此本文總結了問題出現的原因和解決方法,通過這篇文章希望你能解決這個問題。
STAR是一款轉錄組數據的比對軟件,其支持嵌合體的比對方式,也就是說支持一條reads的兩個部分比對到不同的基因組區域,而環狀RNA的junction reads就是符合這樣的要求,代碼如下
STAR \ --genomeDir hg19_star_db/ \ --readFilesCommand gunzip -c \ --readFilesIn R1.fastq.gz R2.fastq.gz \ --runThreadN 4 \ --chimSegmentMin 20 \ --chimScoreMin 1 \ --alignIntronMax 500000 \ --outFilterMismatchNmax 4 \ --alignTranscriptsPerReadNmax 100000 \ --twopassMode Basic \ --outSAMtype BAM SortedByCoordinate \ --chimOutType SeparateSAMold \ --outFilterMultimapNmax 2 \ --outFileNamePrefix C1
雖然軟件提供了一個名為runStar.pl的腳本,但是由于STAR的版本問題,使用起來并不方便。該腳本本質上是對STAR的封裝,直接用STAR就好了,參數設置可以參考腳本中的設置。
第二步就是預測環狀RNA,代碼如下
perl \ postProcessStarAlignment.pl \ --starDir star_out_dir \ --minLen 100 --outDir output_dir
運行完成之后,會三個文件,對應的后綴如下所示
_filteredJunctions.bed
_s_filteredJunctions.bed
_s_filteredJunctions_fw.bed
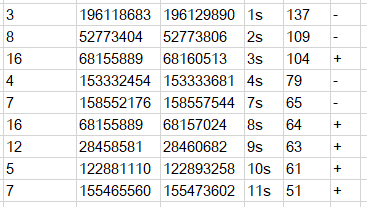
第一個文件為所有環狀RNA的結果文件;第二個文件為剪切位點符合GT-AG剪切信號的環狀RNA;第三個文件和第二個文件的環狀RNA相同,只不過新增了環狀RNA連接點附近的線性RNA平均測序深度信息。通常情況下,我們選擇第二個文件的結果作為最終的環狀RNA預測結果,該文件內容示意如下
看完上述內容,你們掌握circRNA_finder中如何識別環狀RNA的方法了嗎?如果還想學到更多技能或想了解更多相關內容,歡迎關注億速云行業資訊頻道,感謝各位的閱讀!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





