溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這期內容當中小編將會給大家帶來有關如何分析GDC數據庫中的數據的R語言包GDC RNATools,文章內容豐富且以專業的角度為大家分析和敘述,閱讀完這篇文章希望大家可以有所收獲。
GDCRNATools:加利福尼亞大學生物與植物科學系植物基因組學中LNCRNA、miRNA和mRNA數據的綜合分析軟件包
GDC:基因組數據共享
幫助文檔鏈接 http://bioconductor.org/packages/devel/bioc/vignettes/GDCRNATools/inst/doc/GDCRNATools.html
library(GDCRNATools)
project<-'TCGA-CHOL'
rnadir<-paste(project,'RNAseq',sep='/')
mirdir<-paste(project,'miRNAs',sep="/")
gdcRNADownload(project.id = 'TCGA-CHOL',
data.type = 'RNAseq',
write.manifest = F,
method = 'gdc-client',
directory = rnadir)
在linux系統中重復到這一步的時候遇到報錯 ImportError: /lib64/libc.so.6: version `GLIBC_2.18' not found (required by /tmp/_MEIylVP0W/libstdc++
我的解決辦法是把它默認下載的gdc-client_v1.3.0替換掉,我換成gdc-client_v1.5.0,下載地址是https://gdc.cancer.gov/access-data/gdc-data-transfer-tool
gdcRNADownload(project.id = 'TCGA-CHOL',
data.type = 'miRNAs',
write.manifest = F,
method = 'gdc-client',
directory = mirdir)
clinicaldir<-paste(project,'Clinical',sep='/')
gdcClinicalDownload(project.id = 'TCGA-CHOL',
write.manifest = F,
method='gdc-client',
directory = clinicaldir)
metaMatrix.RNA<-gdcParseMetadata(project.id = 'TCGA-CHOL',
data.type = 'RNAseq',
write.meta = F)
metaMatrix.RNA<-gdcFilterDuplicate(metaMatrix.RNA)
metaMatrix.RNA<-gdcFilterSampleType(metaMatrix.RNA)
metaMatrix.MIR<-gdcParseMetadata(project.id = 'TCGA-CHOL',
data.type = 'miRNAs',
write.meta = F)
metaMatrix.MIR
metaMatrix.MIR<-gdcFilterDuplicate(metaMatrix.MIR)
metaMatrix.MIR<-gdcFilterSampleType(metaMatrix.MIR)
rnaCounts<-gdcRNAMerge(metadata = metaMatrix.RNA,
path = rnadir,
organized = FALSE,
data.type = 'RNAseq')
mirCounts<-gdcRNAMerge(metadata = metaMatrix.MIR,
path = mirdir,
organized = FALSE,
rnaCounts[1:5,1:5]
mirCounts[1:5,1:5]
rnaExpr<-gdcVoomNormalization(counts=rnaCounts,filter=F)
mirExpr<-gdcVoomNormalization(counts=mirCounts,filter=F)
rnaExpr[1:5,1:5]
mirExpr[1:5,1:5]
DEGAll<-gdcDEAnalysis(counts = rnaCounts,
group=metaMatrix.RNA$sample_type,
comparison = 'PrimaryTumor-SolidTissueNormal',
method='limma')
deALL<-gdcDEReport(deg=DEGAll,gene.type = 'all')
deLNC<-gdcDEReport(deg=DEGAll,gene.type='long_non_coding')
dePC<-gdcDEReport(deg=DEGAll,gene.type = 'protein_coding')
gdcBarPlot(deg=deALL,angle = 45,data.type = 'RNAseq')
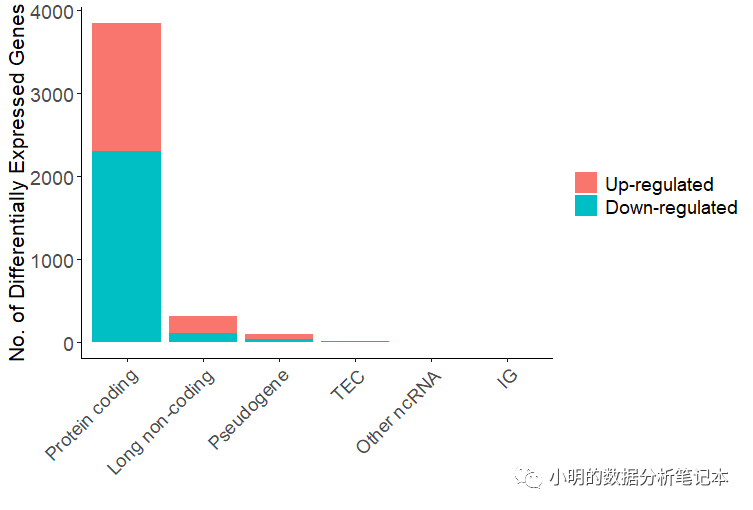
這里TEC和IG分別是啥?
gdcVolcanoPlot(deLNC)
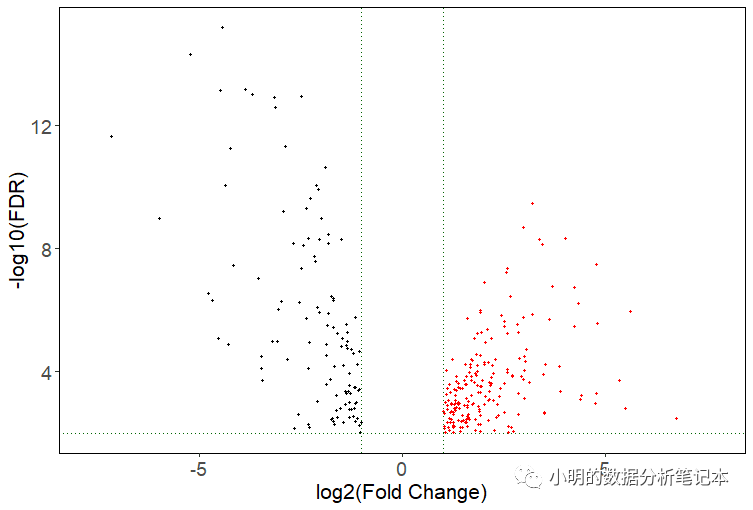
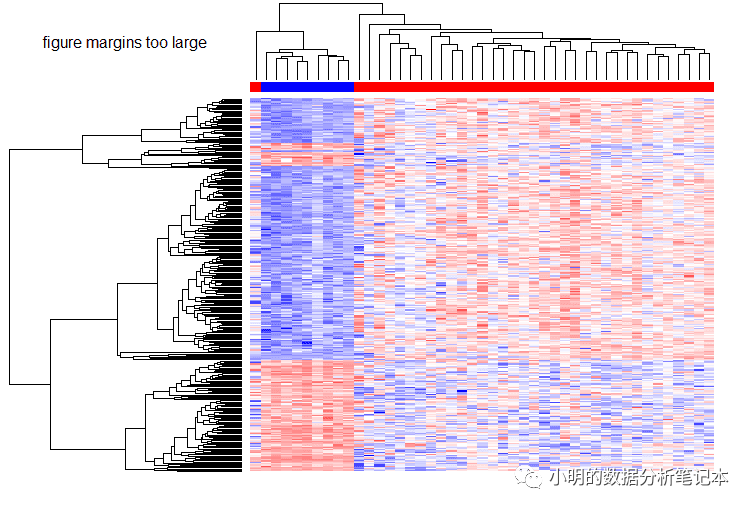
degName<-rownames(deLNC)
gdcHeatmap(deg.id = degName,metadata = metaMatrix.RNA,rna.expr = rnaExpr)
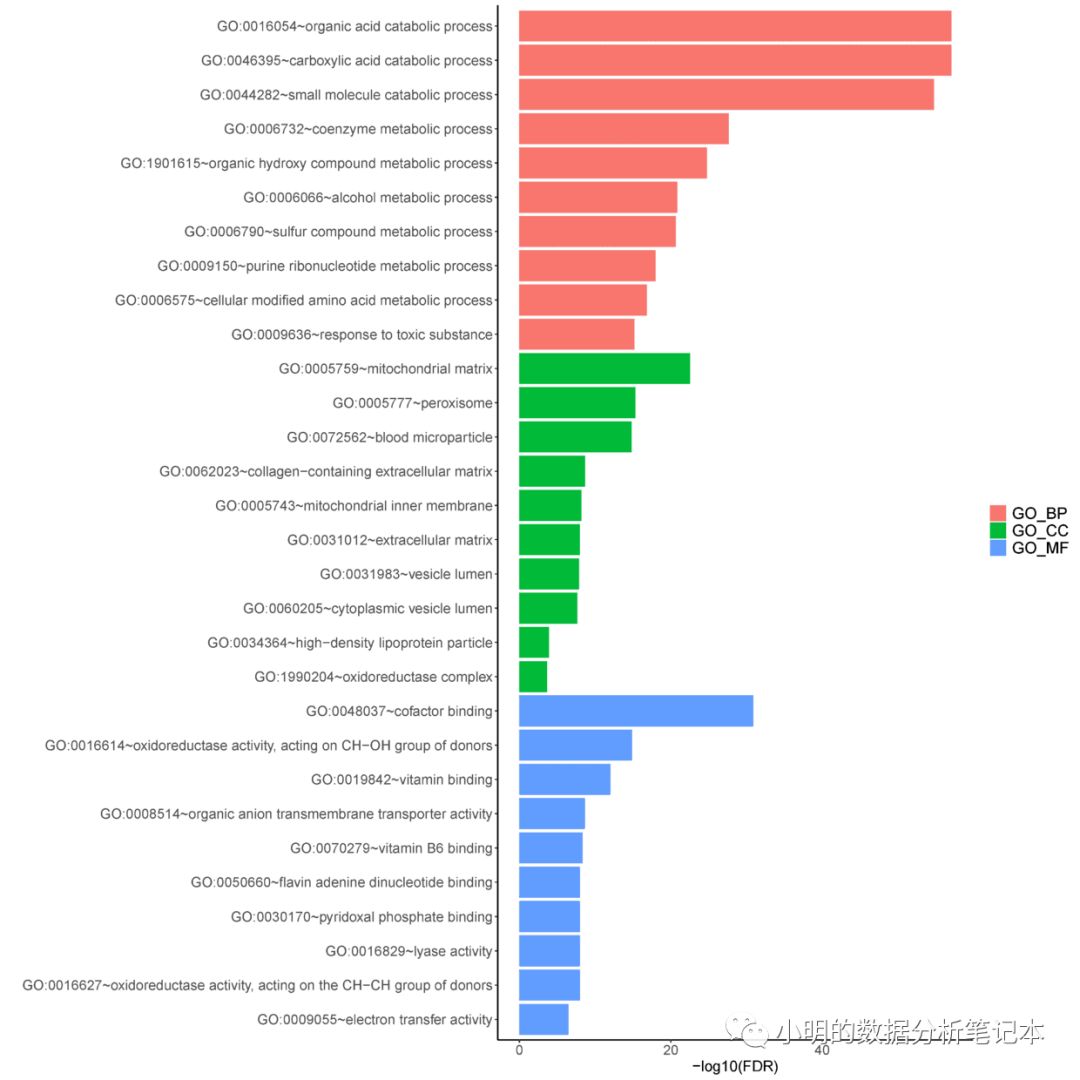
enrichOutput<-gdcEnrichAnalysis(gene=rownames(deALL),
simplify=T)
gdcEnrichPlot(enrichOutput,type='bar',category = 'GO',num.terms = 10)
畫圖的時候遇到報錯 Error in .Call.graphics(C_palette2, .Call(C_palette2, NULL)) : invalid graphics state 不知道原因出在哪里,但是保存到本地沒問題
pdf(file="../goenrich.pdf",width = 15,height = 15)
gdcEnrichPlot(enrichOutput,type='bar',category = 'GO',num.terms = 10)
dev.off()
ceOUtput<-gdcCEAnalysis(lnc=rownames(deLNC),
pc=rownames(dePC),
lnc.targets = 'starBase',
pc.targets = 'starBase',
rna.expr = rnaExpr,
mir.expr = mirExpr)
edges<-gdcExportNetwork(ceNetwork = ceOutput2,net='edges')
nodes<-gdcExportNetwork(ceNetwork = ceOutput2,net='nodes')
write.table(edges,file='edges.txt',sep='\t',quote=F)
write.table(nodes,file="nodes.txt",sep="\t",quote=F)最后生成了兩個文件,如何用cytoscape可視化這兩個文件我暫時還不知道如何實現。
上述就是小編為大家分享的如何分析GDC數據庫中的數據的R語言包GDC RNATools了,如果剛好有類似的疑惑,不妨參照上述分析進行理解。如果想知道更多相關知識,歡迎關注億速云行業資訊頻道。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





