溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
Pheatmap怎樣繪制熱圖,相信很多沒有經驗的人對此束手無策,為此本文總結了問題出現的原因和解決方法,通過這篇文章希望你能解決這個問題。
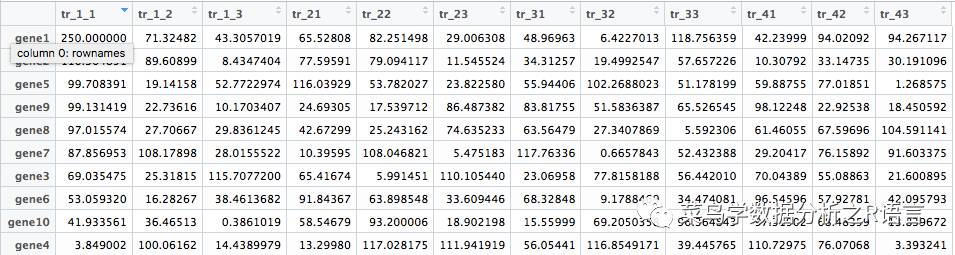
隨機生成,10個基因,每個基因4個處理,每個處理3個平行,表達量RPKM值在1-120之間,矩陣第一個RPKM數值為250:
> library(pheatmap)
> data <- matrix(runif(120,0,120),ncol=12)
> data[1] <- 250
> colnames(data) <- c(paste0('tr_1_',seq(1,3)),paste0('tr_2',seq(1,3)),paste0('tr_3',seq(1,3)),paste0('tr_4',seq(1,3)))
> rownames(data) <- c(paste0('gene',seq(1,10)))
運行過程生成matrix和圖片:
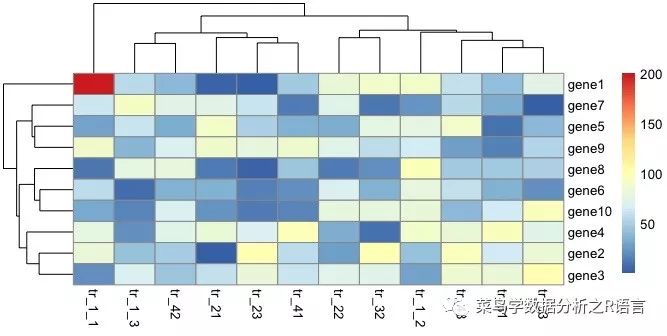
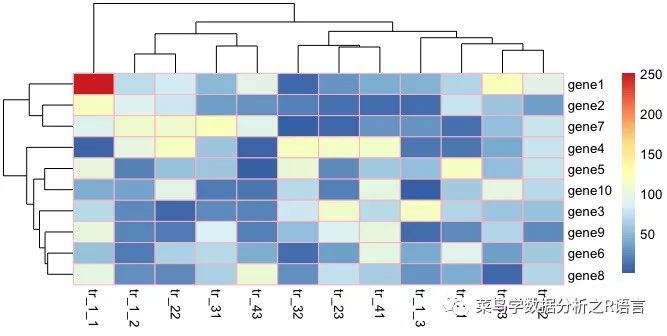
利用border_color參數修改邊界顏色:
>pheatmap(data,border_color = "blue")
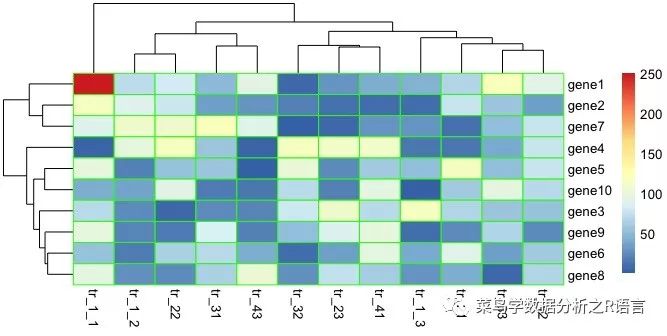
> pheatmap(data,border_color = "red")
> pheatmap(data,border_color = "pink")
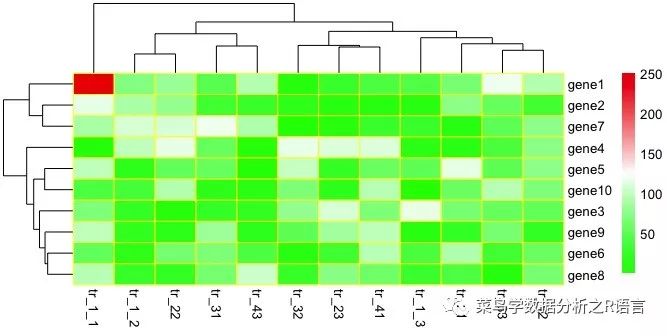
> pheatmap(data,border_color = "green")
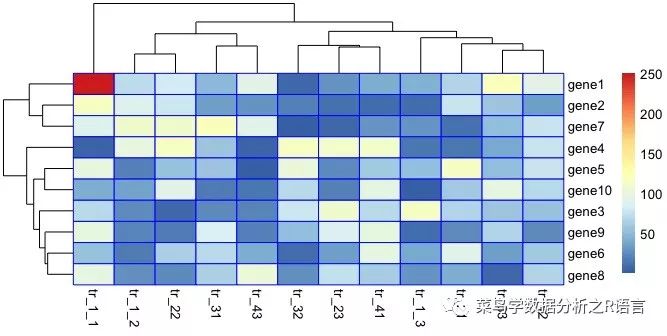
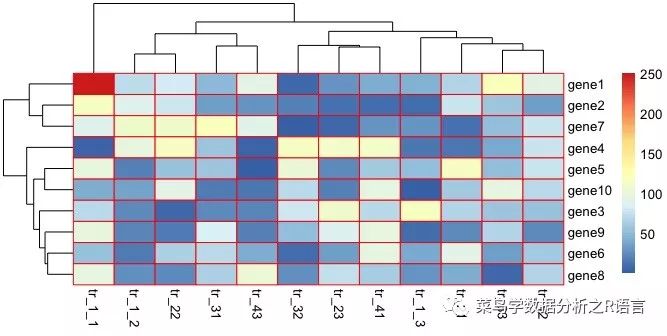
colorRampPalette參數的使用:
> colors <- colorRampPalette(c("blue", "red"))(5)#顏色從藍色到紅色漸變色,5表示長度為5的顏色梯度
> colors
[1] "#0000FF" "#3F00BF" "#7F007F" "#BF003F" "#FF0000"
> pheatmap(data,border_color = "yellow",color=colorRampPalette(c('#00ff00','white','#EE0000'))(100))
運行結果如下:
scale參數的使用:
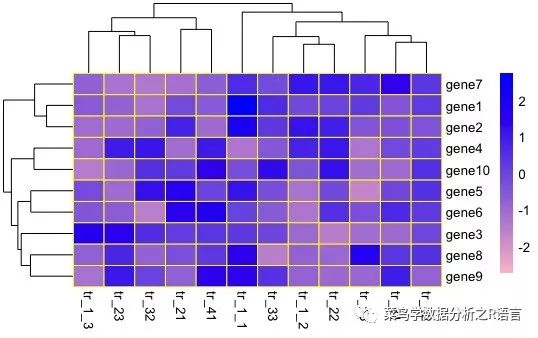
scale是指對數值進行均一化處理,在基因表達量的數據中,有些基因表達量極低,有些基因表達量極高,因此把每個基因在不同處理和重復中的數據轉換為平均值為0,方差為1的數據,可以看出每個基因在某個處理和重復中表達量是高還是低。

“row”、“colume”、“none”分別表示對成行或成列的進行均一化,或不做均一化,一般數據處理中基因的表達量做均一化處理,選擇“row”
> pheatmap(data,border_color = "yellow",color = colorRampPalette(c('pink','blue'))(100),scale='row')
參數:cluster_rows/cluster_cols和cellwidth/cellheight
對基因的順序進行聚類,因此可使用cluster_rows/cluster_col來修改;同時可以使用cellwidth/cellheight對每個單元方塊的大小進行設置:
>pheatmap(data,border_color='yellow',color=colorRampPalette(c('pink','blue'))(100),scale='row',cluster_cols = FALSE,cellwidth = 20,cellheight = 20)
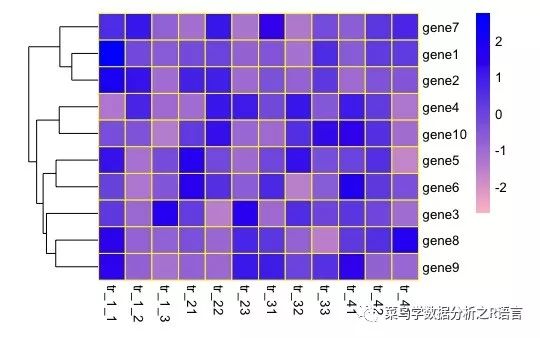
參數:legend/legend_breaks/legend_labels
使用legend閾值邏輯值來對色度條進行隱藏,以及對色度條上對應位置的字符進行修改:
>pheatmap(data,border_color='yellow',color=colorRampPalette(c('pink','blue'))(100),scale='row',cluster_cols = FALSE,cellwidth = 20,cellheight = 20,legend_breaks = c(-2.5,0,2.5),legend_labels = c('>=-2.5','0','2.5'))
運行結果如下:
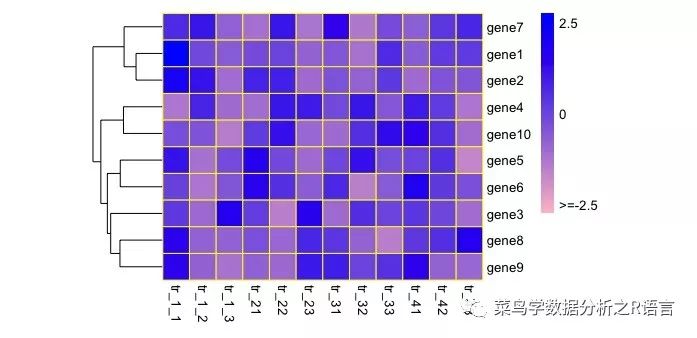
>pheatmap(data,border_color='yellow',color=colorRampPalette(c('pink','blue'))(100),scale='row',cluster_cols = FALSE,cellwidth = 20,cellheight = 20,legend=FALSE,legend_breaks = c(-2.5,0,2.5),legend_labels = c('>=-2.5','0','2.5'))
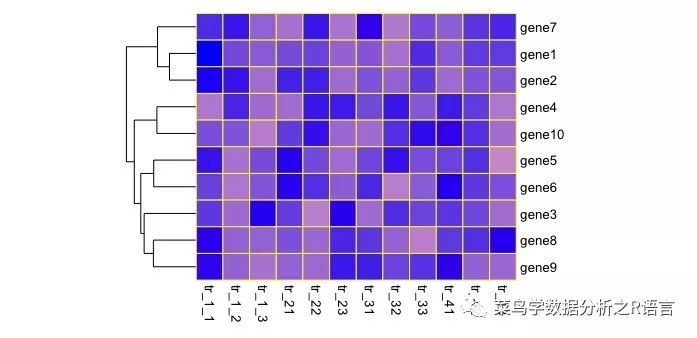
參數:gaps_row/gaps_col、cutree_rows/cutree_cols和treeheight_row/treeheight_col
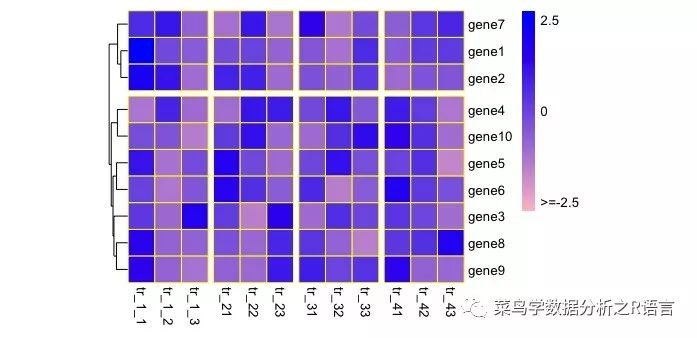
cutree_rows按聚類分割,如cutree_rows=2,把基因表達量聚類分成2類;gaps_col=c(3,6,9)不能聚類,把重復都分開。gaps_XX對行或列進行分割,就不應對相應的行或列進行聚類;treeheight_row參數改變聚類的支長長度:
>pheatmap(data,border_color='yellow',color=colorRampPalette(c('pink','blue'))(100),scale='row',cluster_cols = FALSE,cellwidth = 20,cellheight = 20,legend=FALSE,legend_breaks = c(-2.5,0,2.5),legend_labels = c('>=-2.5','0','2.5'),cutree_rows = 2,gaps_col = c(3,6,9),treeheight_row = 10)
參數:annotation_row/annotation_col、annotation_colors、annotation_legend和annotation_names_row/annotation_names_col
利用annotation_col參數,給各個處理添加一個顏色標簽;
利用annotation_colors對標簽的顏色進行修改;
利用annotation_legend設置是否顯示標簽注釋條;
利用annotation_names_col設置是否顯示標簽名稱。
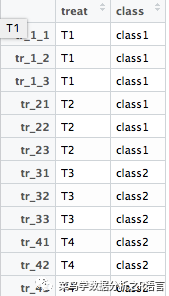
>annotation_col=data.frame(treat=factor(rep(paste0('T',1:4),each=3)),class=factor(rep(paste0('class',1:2),each=6)))
>ann_color=list(a=c(T1='yellow',T2='#757083',T3='firebrick',T4='#66A61E'),b=c(class1='blue',class2='#1B9E77'))
> row.names(annotation_col)=colnames(data)
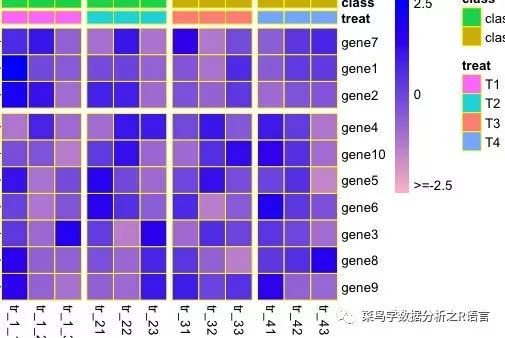
>pheatmap(data,border_color='yellow',color=colorRampPalette(c('pink','blue'))(100),scale='row',cluster_cols = FALSE,cellwidth = 20,cellheight = 20,legend_breaks = c(-2.5,0,2.5),legend_labels = c('>=-2.5','0','2.5'),cutree_rows = 2,gaps_col = c(3,6,9),treeheight_row = 10,annotation_col=annotation_col,annotation_legend=TRUE,annotation_colors=ann_color,annotation_names_col=TRUE)
運行過程中產生數據與圖:
annotation_col:
參數:display_numbers、number_format、number_color和fontsize_number
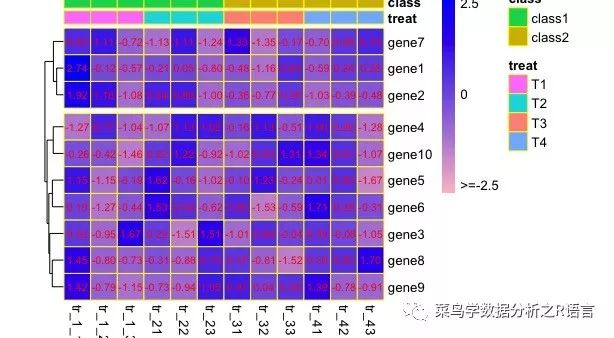
利用display_numbers參數可以在每個單元框內顯示每個方框對于的數據,其中有三個選項,TRUE、FALSE以所對應的數據,如果設置display_numbers=T,這顯示做了均一化的數據(如果之前使用過scale參數),設置display_numbers=data,則表示為直接顯示原始數據,即可直接顯示出RPKM值在單元格中;
number_color顧名思義就是這是設置顯示數據的顏色了
fontsize_number則為顯示每個數據的大小;
利用number_format可以設置保留小數位數或者字符串格式(如%.2f),但僅有在display_numbers=T時才能使用,很雞肋,因此不建議使用該參數,而我們一般是直接顯示RPKM值,所以我們需要之前對數據集進行保留小數處理,不然數據顯示會超出單元格
>pheatmap(data,border_color='yellow',color=colorRampPalette(c('pink','blue'))(100),scale='row',cluster_cols = FALSE,cellwidth = 20,cellheight = 20,legend_breaks = c(-2.5,0,2.5),legend_labels = c('>=-2.5','0','2.5'),cutree_rows = 2,treeheight_row = 10,annotation_col=A,annotation_legend=TRUE,annotation_colors=ann_color,annotation_names_col=TRUE,display_numbers = TRUE,number_color = 'red',fontsize_number = 8,number_format = "%.2f")
運行結果如下:
 參數:show_rownames/show_colnames、fontsize_col/fontsize_row、fontsize和main
參數:show_rownames/show_colnames、fontsize_col/fontsize_row、fontsize和main
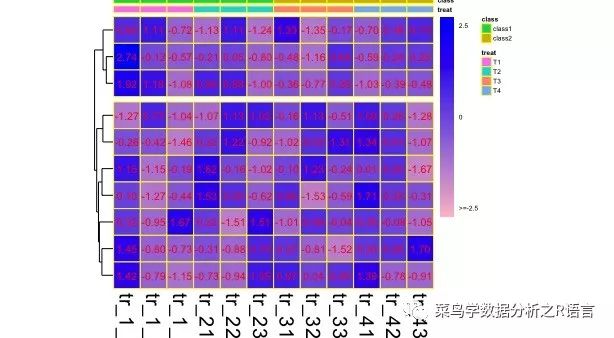
show_rownames表示是否顯示gene名稱,用邏輯值設置,fontsize_col設置橫坐標名稱的大小,fontsize則是設置所有除主圖以外的標簽的大小,利用main設置熱圖的名稱,如:
>pheatmap(data,border_color='yellow',color=colorRampPalette(c('pink','blue'))(100),scale='row',cluster_cols = FALSE,cellwidth = 20,cellheight = 20,legend_breaks = c(-2.5,0,2.5),legend_labels = c('>=-2.5','0','2.5'),cutree_rows = 2,treeheight_row = 10,annotation_col=A,annotation_legend=TRUE,annotation_colors=ann_color,annotation_names_col=TRUE,display_numbers = TRUE,number_color = 'red',fontsize_number = 8,number_format = "%.2f",show_rownames = FALSE,fontsize_col = 15,fontsize=5,main = "heatmap test 2")
看完上述內容,你們掌握Pheatmap怎樣繪制熱圖的方法了嗎?如果還想學到更多技能或想了解更多相關內容,歡迎關注億速云行業資訊頻道,感謝各位的閱讀!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





