溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
TCGA數據庫的normal樣本不夠該怎么辦,相信很多沒有經驗的人對此束手無策,為此本文總結了問題出現的原因和解決方法,通過這篇文章希望你能解決這個問題。
自己想挖掘的癌癥,雖然是在TCGA數據庫有數據,但是normal(癌旁樣品或者血液)太少了,做差異分析什么的, 會面臨樣本數量不平衡問題,是否可以納入GTEx數據庫的正常組織轉錄組測序數據。
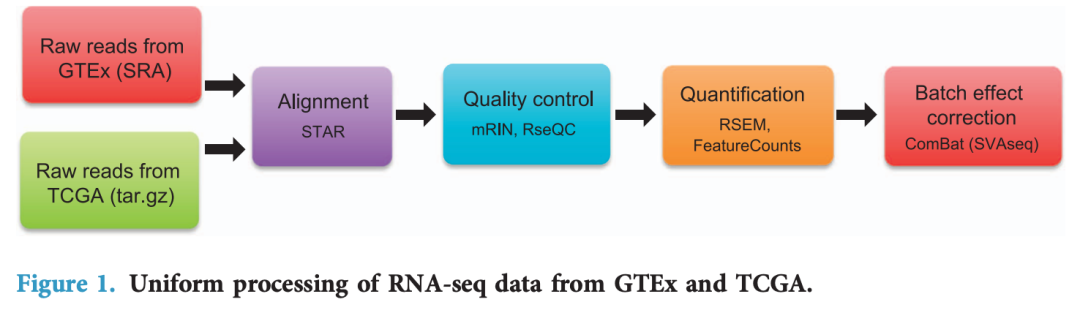
其實是沒辦法簡單的回答是否可以整合TCGA和GTEx數據庫,或者說該如何結合,這背后的統計學略微有點復雜,不僅僅是批次效應。發表在Sci Data. 2018; 的文章:Unifying cancer and normal RNA sequencing data from different sources 就比較詳細的說明了TCGA和GTEx數據庫的轉錄組數據的天然差異:
全部代碼共享在:GitHub (https://github.com/mskcc/RNAseqDB).
最近一篇發表在SR,17 February 2020 的文章:Variability in estimated gene expression among commonly used RNA-seq pipelines 比較了常見轉錄組測序數據分析流程對定量拿到的表達矩陣的影響:
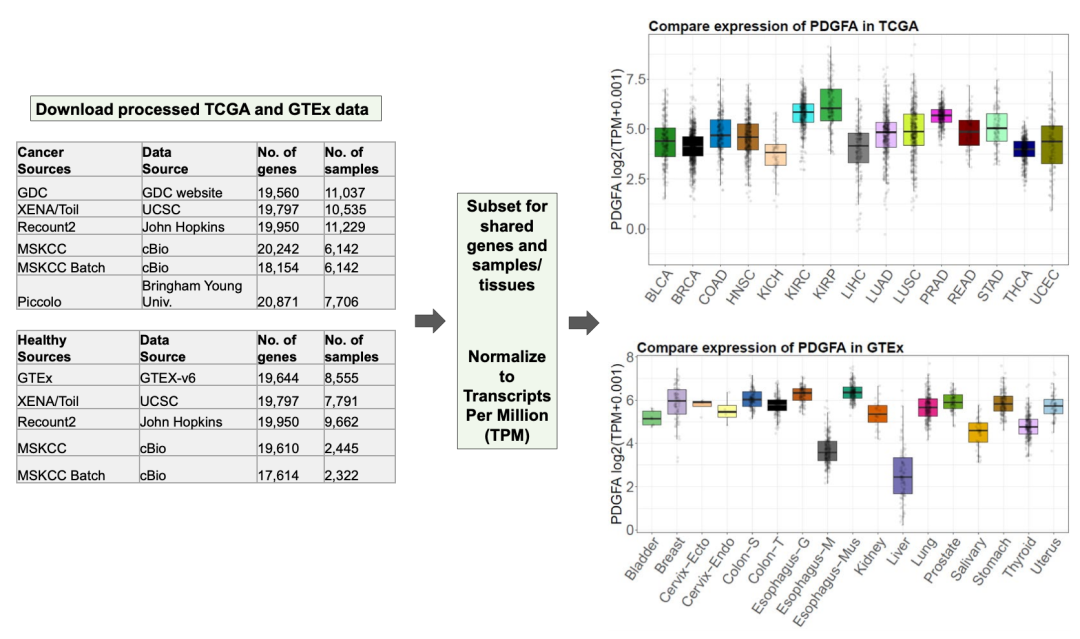
TCGA和GTEX是兩個超級大的擁有RNA-seq數據的計劃,其中TCGA涵蓋33種癌癥,超1萬個樣品,而GTEX也有500多個病人的50多種組織的近1萬個樣品數據。它們各自的發起單位對RNA-seq數據處理不一樣,而且后續也有一些新的流程處理試圖統一兩個數據庫的RNA-seq數據分析結果,比較出名的5個流程分別是:
作者把這5個流程應用到TCGA和GTEX,得到10個不同組合的數據
做了非常完善的比較,并且公布全部代碼在:https://github.com/sonali-bioc/UncertaintyRNA
非常多!
很多簡陋的數據挖掘,比如發表在PeerJ的 BIOINFORMATICS AND GENOMICS雜志的文章:Identification of four hub genes associated with adrenocortical carcinoma progression by WGCNA 也會涉及到TCGA數據庫和GTEx的整合。
首先下載TCGA和GTEx數據庫的TPM表達矩陣:
Gene transcripts per million (TPM) data were downloaded from the UCSC Xena database, which included ACC (The Cancer Genome Atlas, n = 77) and normal samples (Genotype Tissue Expression, n = 128).
然后差異分析流程是:
Of the 60,498 genes in each sample, we removed genes with a mean TPM ≤ 2.5 (>1 is a common cutoff for determining if an isoform is expressed or not in the cancer and normal samples and thus retained 13,987 genes.
For those genes in the samples that showed significant changes, we used analysis of variance (ANOVA) in R to determine the variance in genes between the two groups. ANOVA is a collection of statistical models useful for DEG analysis.
We obtained 2,953 significant DEGs (Table S2) in ACC with a p < 0.001 and |log2 (fold-change)| > 1 cutoff.
差異分析結果是:1,181 up-regulated and 1,772 down-regulated genes.
可以看到,作者默認TPM這個轉錄組測序表達數據歸一化形式本身是具有跨平臺跨數據庫的特性,所以無需考慮批次效應,直接使用最簡單粗暴的ANOVA檢驗即可!
我們都知道,TCGA數據庫是目前最綜合最全面的癌癥病人相關組學數據庫,包括:
知名的腫瘤研究機構都有著自己的TCGA數據庫探索工具,比如:
對轉錄表達這個層面的信息來說,最優選擇當然是整合TCGA和GTEx數據庫,但是對于甲基化數據,我們有沒有類似于GTEx數據庫的超級大隊列呢?
目前我還沒有接觸到,我前面分享過:這樣的診斷模型才優秀,作者就是下載TCGA的結直腸癌甲基化位點信號矩陣文件:
以及正常人的血液的甲基化信號值作為對照:
上面的兩個隊列是為了確定直腸癌特異性甲基化位點,做的是差異分析,確定了 top 1000 methylation markers
可以合理的推測應該是沒有人類各個正常組織的甲基化數據供使用,所以他們才會退而求其次使用正常人的血液的甲基化信號值作為對照吧!
看完上述內容,你們掌握TCGA數據庫的normal樣本不夠該怎么辦的方法了嗎?如果還想學到更多技能或想了解更多相關內容,歡迎關注億速云行業資訊頻道,感謝各位的閱讀!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





