溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
今天就跟大家聊聊有關如何進行MACS2 peak calling的實戰,可能很多人都不太了解,為了讓大家更加了解,小編給大家總結了以下內容,希望大家根據這篇文章可以有所收獲。
MACS是一款最為流行的peak calling軟件,最初是針對轉錄因子的chip數據來設計的,在最新版本中,也添加了對組蛋白修飾的適配。目前最新版本為v2.0,官網如下
https://github.com/taoliu/MACS
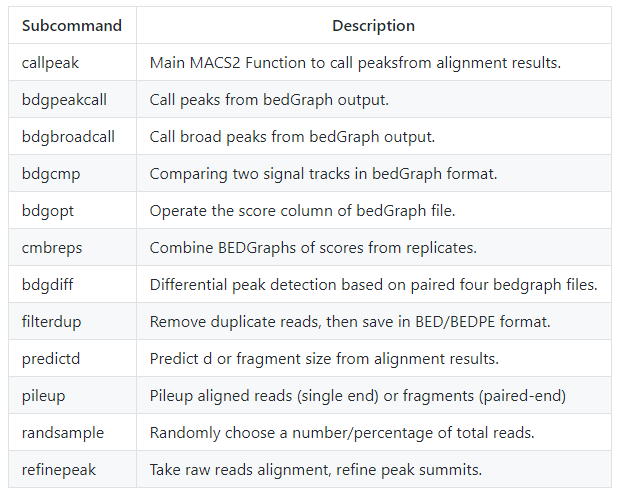
在2.0版本中提供了以下多個子命令
callpeak
bdgpeakcall
bdgbroadcall
bdgcmp
bdgopt
cmbreps
bdgdiff
filterdup
predictd
pileup
randsample
refinepeak
每個子命令和對應的功能描述如下
下面主要介紹macs2最經典的使用場景peak calling, 基本用法如下
macs2 callpeak \
-t ip.bam \
-c input.bam \
--outdir out_dir \
-n chip \
-g hs-t參數指定抗體處理的樣本,-c指定input樣本,值得一提的是,macs支持多種格式的輸入文件,除了上述代碼中使用的bam格式外,還支持SAM/BED格式。
--outdir指定輸出結果的目錄,-n參數指定輸出文件名的前綴,-g參數指定基因組的有效大小,在NGS數據中,測序reads在基因組上的覆蓋度并不是100%, 而且有些重復區域的比對信息是不可信的,剩下的能夠利用的區域通常只占整個基因組區域的70%到90%,這個區域的大小就是有效大小,對于常見的物種,程序內置了有效大小,我們只需要指定物種的縮寫即可

對于其他物種,則需要自己指定有效基因組的大小,單位為bp。
輸出文件如下
chip_model.r
chip_peaks.narrowPeak
chip_peaks.xls
chip_summits.bedmodel.r是一個可執行的R腳本,通過以下代碼可以產生一個PDF的輸出文件
Rscript chip_model.r
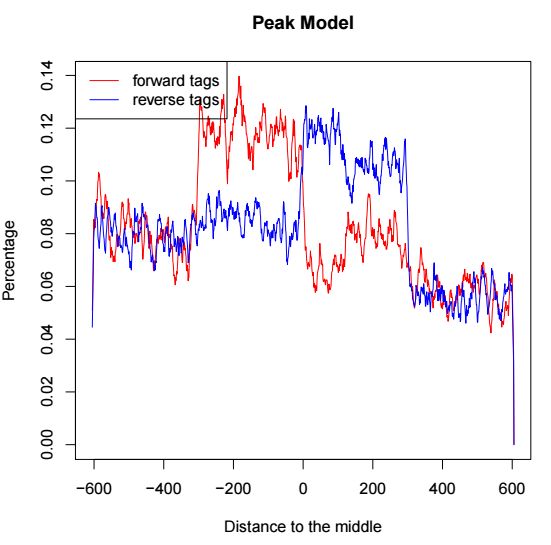
第一頁表示peak鄰近區間正負鏈測序分布,用于評估d這個參數值,示意如下
第二頁是cross-correlation分析的結果,示意如下

后綴為xls的文件是peak的輸出結果,內容示意如下
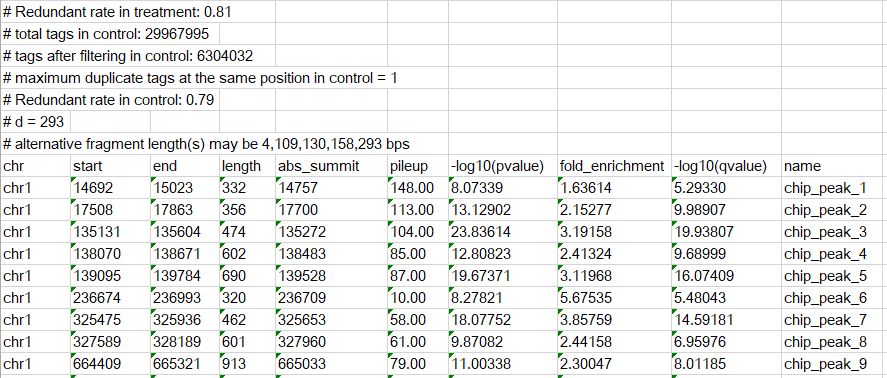
#開頭的是注釋信息,顯示了軟件調用的具體命令和參數設置,便于核查;其他的行記錄了peak的區間信息,這里的起始位置采用的是從1開始計數的方式。
后綴為narrowpeak的文件是一個BED格式的文件,內容示意如下
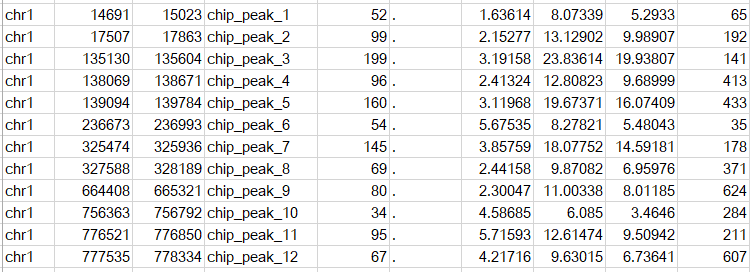
前四列代表peak區間和名稱,注意bed格式中起始位置從0開始計數,第五列的值為int(-10*log10qvalue),第六列全部為.,第七列為fold_enrichment,第八列為-log10(pvalue),第九列為-log10(qvalue),第十列為peak的中心,即summit距離peak起始位置的距離,對應abs_summit - start。
后綴為bed的文件為peak中心,即summit對應的bed文件,內容示意如下
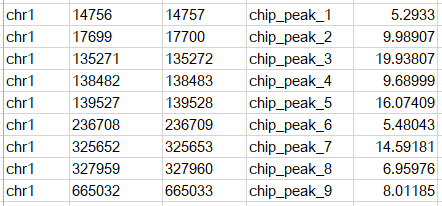
最后一列為-log10(qvalue)。
看完上述內容,你們對如何進行MACS2 peak calling的實戰有進一步的了解嗎?如果還想了解更多知識或者相關內容,請關注億速云行業資訊頻道,感謝大家的支持。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





