溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這篇文章主要介紹“如何使用deeptools查看reads分布特征”,在日常操作中,相信很多人在如何使用deeptools查看reads分布特征問題上存在疑惑,小編查閱了各式資料,整理出簡單好用的操作方法,希望對大家解答”如何使用deeptools查看reads分布特征”的疑惑有所幫助!接下來,請跟著小編一起來學習吧!
在chip_seq數據分析中,通常會對peak區域在基因組上的分布進行探究,查看其分布是否存在規律,比如是否在轉錄起始位點,或者轉錄終止位點附近存在富集,此時我們可以通過deeptools這個工具來實現。
首先通過computeMatrix這個命令,可以計算基因組區域上的分布,分成以下兩種模式
scale-regions
reference-point
第一種模式代表一個區間,包含了起始和終止位置,第二種模式代表的是某一個位點,比如轉錄起始位點。對于這兩個模式的區別,官網給出了很好的解釋,示意如下
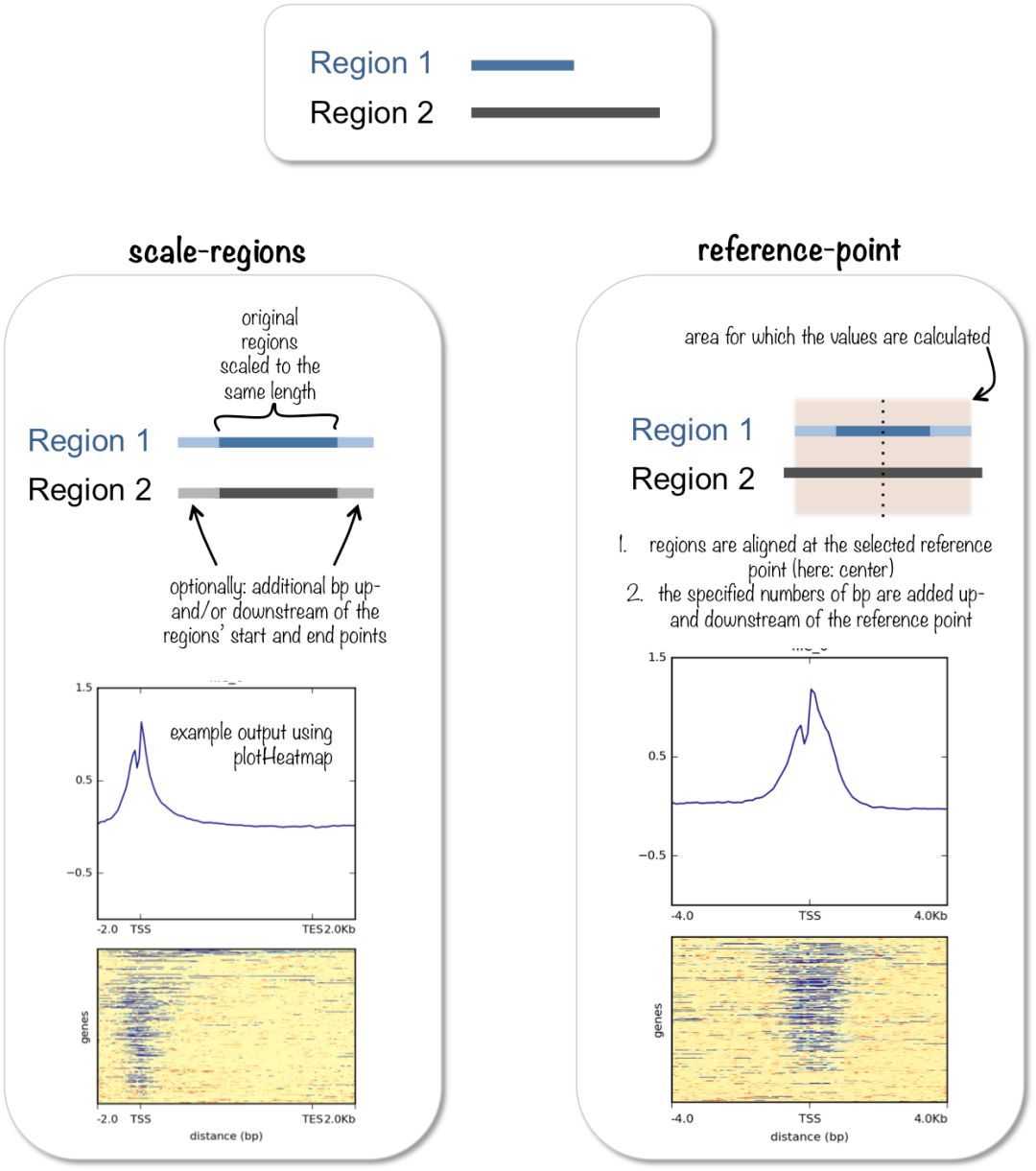
將所有的區域劃分為等長的區間稱之為bin, 然后計算每個bin內所有位點的測序深度,默認用所有位點測序深度的平均值來代表這個區間。通過這個命令計算得到中間結果文件之后,可以使用以下兩個命令進行可視化
plotProfile
plotHeatmap
下面展示一個實際的例子,從bam文件開始,得到最終的可視化結果
通過bamCoverage命令,可以將bam文件轉換為bigwig文件,用法如下
bamCoverage -b input.bam -o input.bw
這個命令有scale-regions和reference-points兩種模式,這里以第二種為例進行展示,用法如下
computeMatrix reference-points \
-S inpnut.bw \
-R hg19.bed \
--binSize 10 \
--skipZeros \
-a 3000 \
-b 3000 \
-o matrix.gz \
--outFileNameMatrix matrix.tab在輸出的tab文件中,每一行代表一個轉錄本,和輸入的bed文件中的轉錄本個數一致,每一列代表bin區間內的平均測序深度,列數的多少和區間的長度以及bin_sizz有關。在上面這個例子中,選擇上下游各3kb的區間,bin大小為10bp, 所以總共有3000X2/10, 即600個區間。
在可視化時,區間個數越多,畫出來的折線圖會相對平滑,所以可以適當調整bin的大小,使畫出來的圖更加美觀。
用法如下
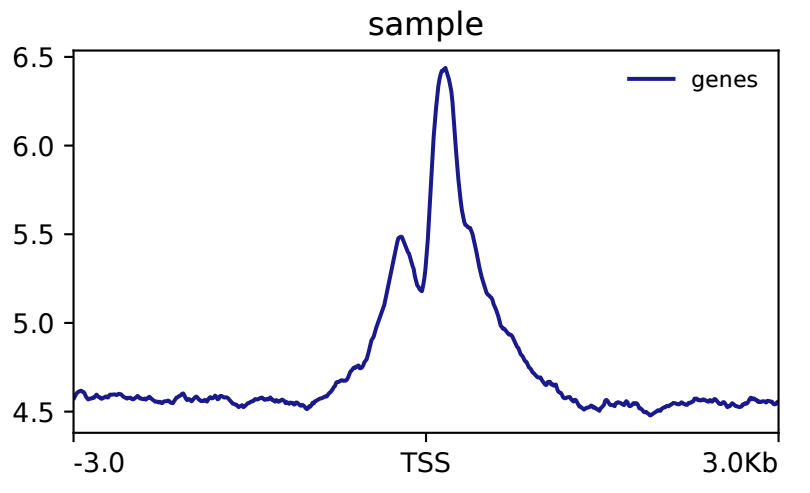
plotProfile -m matrix.gz \
-out profile.pdf生成的結果如下
用法如下
plotHeatmap -m matrix.gz -o heatmap.pdf
生成的結果分成了兩部分,第一部分和plotProfile的結果相同,第二部分是一個熱圖,示意如下
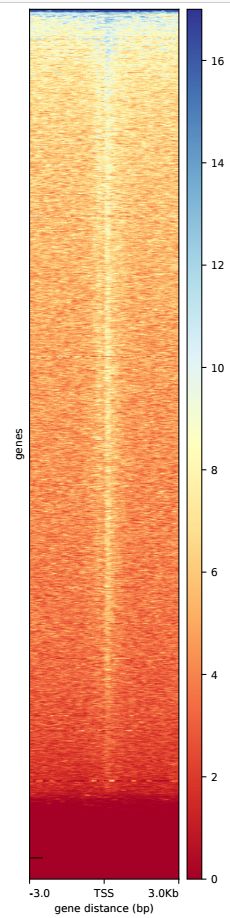
就是將生成的tab文件中的內容繪制了一個熱圖,以上展示的都是基本用法,除此之外,還有很多的參數可以調整,繪制出更加美觀的圖片。
到此,關于“如何使用deeptools查看reads分布特征”的學習就結束了,希望能夠解決大家的疑惑。理論與實踐的搭配能更好的幫助大家學習,快去試試吧!若想繼續學習更多相關知識,請繼續關注億速云網站,小編會繼續努力為大家帶來更多實用的文章!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





