溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
本篇文章為大家展示了methylKit是進行差異甲基化分析,內容簡明扼要并且容易理解,絕對能使你眼前一亮,通過這篇文章的詳細介紹希望你能有所收獲。
methylKit 是一個用于分析甲基化測序數據的R包,不僅支持WGBS,RRBS和目的區域甲基化測序,還支持oxBS-sq, TAB-seq等分析5hmc的數據。 其核心功能是差異甲基化分析和差異甲基化位點和區域的注釋。
安裝過程如下:
source(“http://bioconductor.org/biocLite.R“)
biocLite(“methylKit”)
推薦使用最新版本的R進行安裝,這樣可以使用最新版本的methylKist。
利用methylKit 做差異分析包括3步
每個樣本一個原始數據,methylKit支持兩種格式的methylation calling文件
純文本格式
內容如下
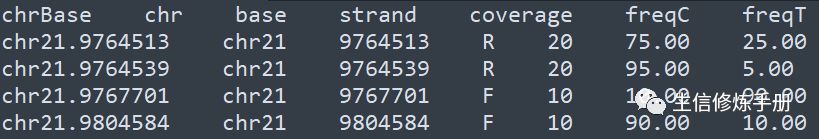
每一行是一個甲基化位點,coverage 代表覆蓋這個位點的reads數,freqC 代表甲基化C的比例,freqT 代表非甲基化C的比例。這種純文本格式內容非常直觀,文件大小相比bam 文件小很多,讀取的速度更快。
純文本格式的讀取過程如下
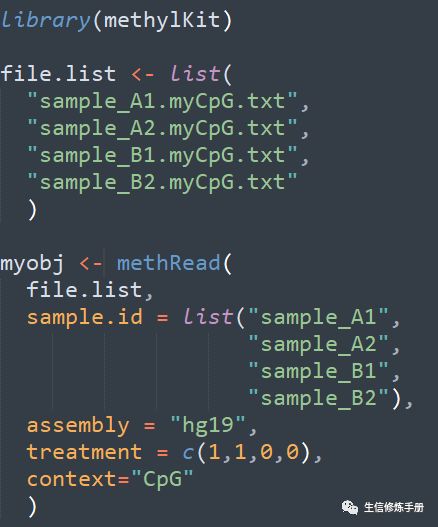
treatment參數指定樣本的分組,0代表control組,1代表treatment組
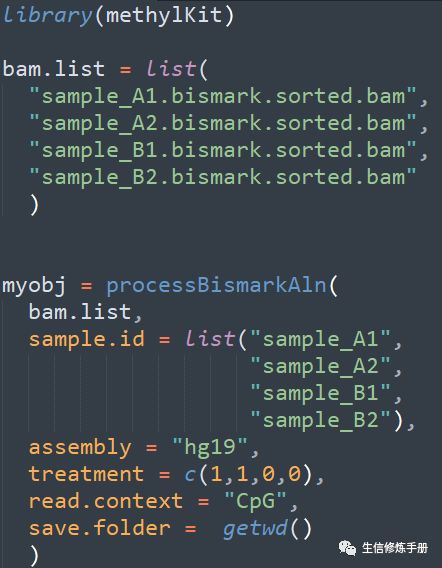
bam文件
直接讀取Bismark軟件比對產生的bam文件,通過processBismarkAln實現
用法如下:
將所有樣本的甲基化情況合并,得到所有樣本的甲基化表達譜,用法如下
meth=unite(myobj, destrand=FALSE)
meth 中的內容如下,其實就是之前的methylation calling文件的合并
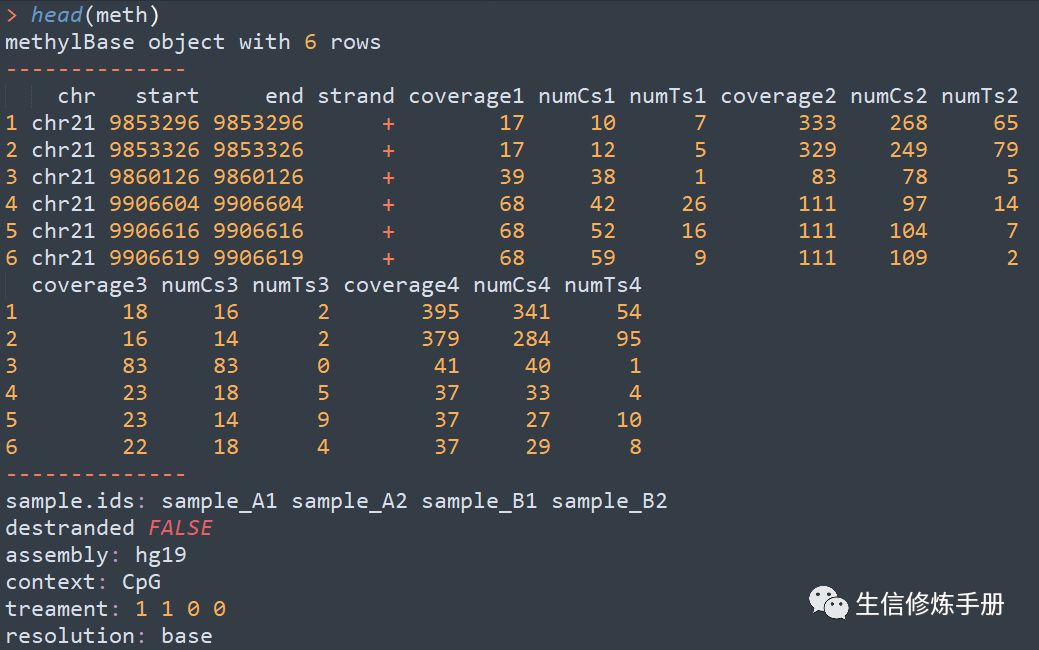
在合并的過程中,默認情況下,只有所有的樣本都包含該位點時,才會保留,本質就是取的所有樣本的交集,如果你想要取并集,可以修改min.per.group參數的值,該參數的值代表每組中至少有多少個樣本覆蓋該位點時才保留,如果設置為1,就是取并集。
meth.min=unite(myobj,min.per.group=1L)
通過calculateDiffMeth函數來執行差異甲基化分析,用法如下
myDiff=calculateDiffMeth(meth)
根據甲基化C是變多了還是變少了,可以將差異甲基化的結果分為兩大類:
hypermethylated
hypomethylated
hypermethylated表示相比control組,treatment組中的甲基化C更多;hypomethylated則相反,表示treatment組中的甲基化C比control組中少。
采用getMethylDiff函數提取差異分析的結果,用法如下
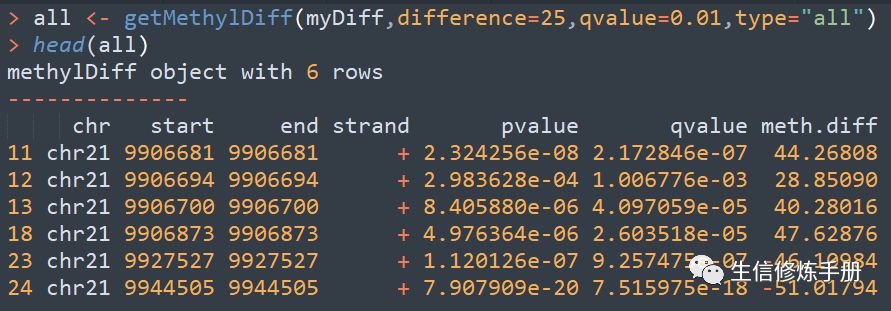
difference函數表明差異的閾值,只有差異大于該閾值時,才會保留,起始就是meth.diff的值,注意是絕對值大于difference的值。
除了difference閾值之外,還有qvalue閾值,小于該閾值的結果保留。在methylKit中,校正p值采用的是SILM算法,和我們常規的BH算法不同。type參數定義差異的類型,如果你只關注hypermethylated或者hypomethylated,可以設置type 參數的值,單獨篩選。
在methylKit中,它的差異分析總是針對合并后的甲基化表達譜,如果你的甲基化表達譜每一行是一個甲基化位點,那么差異分析的結果就是差異甲基化位點;如果你的表達譜每一行是一個甲基化區域,那么差異分析的結果就是差異甲基化區域。上面的例子都是針對差異甲基化位點的,下面看下差異甲基化區域的分析。
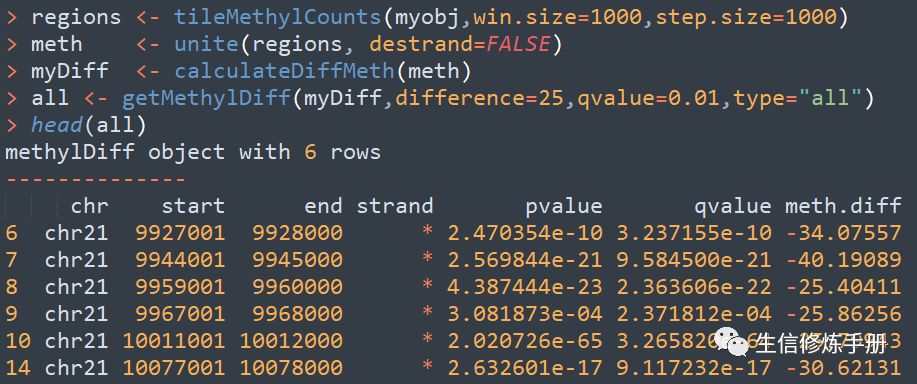
首先遇到的問題就是甲基化區域如何界定,在methylKit中,按照滑動窗口的方式定義甲基化區域,默認窗口大小為10000 bp ,步長為10000bp,通過tileMethylCounts函數實現。
完整的差異甲基化區域分析的代碼如下:
上述內容就是methylKit是進行差異甲基化分析,你們學到知識或技能了嗎?如果還想學到更多技能或者豐富自己的知識儲備,歡迎關注億速云行業資訊頻道。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





