溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
小編給大家分享一下如何實現bigwig歸一化,相信大部分人都還不怎么了解,因此分享這篇文章給大家參考一下,希望大家閱讀完這篇文章后大有收獲,下面讓我們一起去了解一下吧!
在chip_seq數據展示時,經常會用到bigwig文件,導入igvtools等基因組瀏覽器中,產生如下所示的圖片
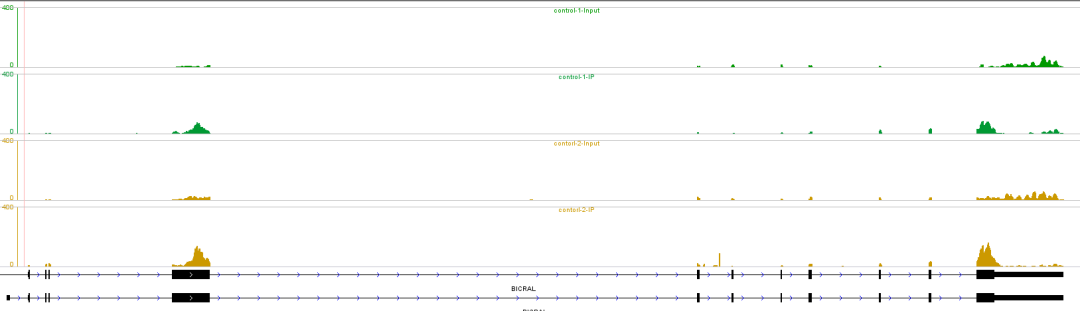
我們將IP樣本相對Input樣本中reads富集的區域定義為peak, 反映到上圖中,則對應的為IP樣本中reads出現了峰值,比如下圖紅色標記的區域
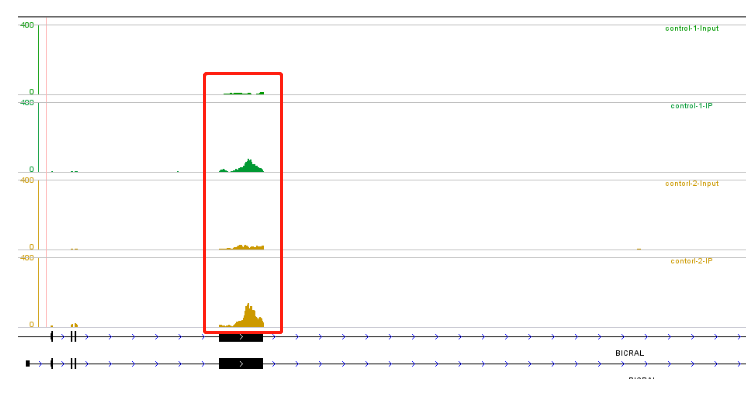
通過這種可視化的方式,可以直觀的反映出peak區域的情況,但是在實際使用中需要注意歸一化的問題。
bigwig文件本質上展示的是測序深度的分布信息,而原始的測序深度是和測序的reads量呈正相關關系的,比如Input樣本測序5G, IP樣本測序10G, 在原始的測序深度看,會看到Input樣本相比IP樣本,其測序深度是偏高的。當然這個是一個極端的例子,但是很好的說明了測序量的差異對原始的測序深度會有直接的影響。
為了消除樣本間測序數據量差異的影響,很當然的我們想到了歸一化,類似轉錄組中的定量策略,原始的測序深度就是raw count, 那么當然類似RPKM, CPM等歸一化方式,對于bigwig文件而言,同樣適用。
在deeptools中,提供了多種歸一化方式
RPKM的公式如下
RPKM (per bin) = number of reads per bin / (number of mapped reads (in millions) * bin length (kb))
用法如下
deeptools bamCoverage \
-p 10 \
--bam input.bam \
--normalizeUsing RPKM \
--outFileName rpkm.bigwigCPM的公式如下
CPM (per bin) = number of reads per bin / number of mapped reads (in millions)
用法如下
deeptools bamCoverage \
-p 10 \
--bam input.bam \
--normalizeUsing CPM \
--outFileName cpm.bigwigBPM的公式如下
BPM (per bin) = number of reads per bin / sum of all reads per bin (in millions)
用法如下
deeptools bamCoverage \
-p 10 \
--bam input.bam \
--normalizeUsing BPM \
--outFileName bpm.bigwigRPGC的公式如下
RPGC (per bin) = number of reads per bin / scaling factor for 1x average coverage
scaling factor = (total number of mapped reads * fragment length) / effective genome size用法如下
deeptools bamCoverage \
-p 10 \
--bam input.bam \
--normalizeUsing RPGC \
--effectiveGenomeSize 2864785220 \
--outFileName rpgc.bigwig對于同一個樣本而言,導入igvtools中,幾種歸一化方式產生的bigwig文件和原始的bigwig文件的峰形是完全一樣的 ,示意如下
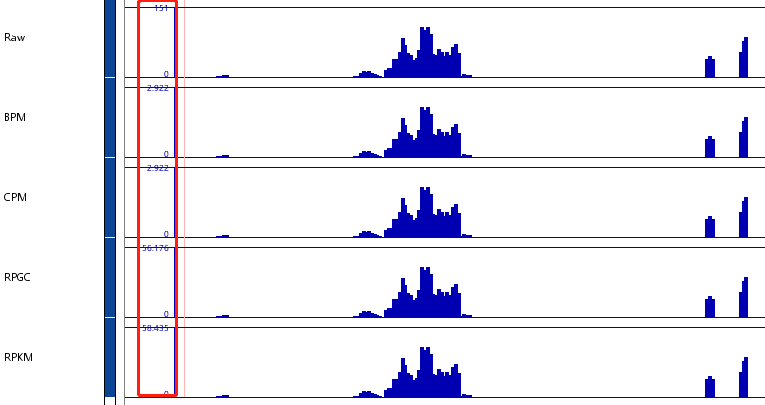
注意紅色方框標記的縱軸的范圍,可以看到不同方式,其縱軸范圍不一樣。
歸一化主要用于樣本間的比較, 比如在比較Input和Ip兩個樣本時,就應該使用歸一化之后的數據,以RPKM為例,導入之后可以看到如下所示的結果
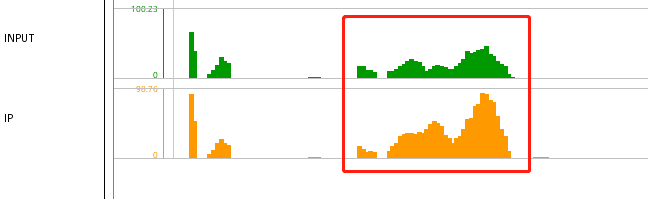
可以看到縱軸的范圍是不一致的,為了更好的比較樣本間的差異,我們需要把二者的縱軸范圍調整成一致的,因為數據已經做了歸一化處理,所以可以直接在同一范圍內進行比較,設置成同一范圍后,效果如下
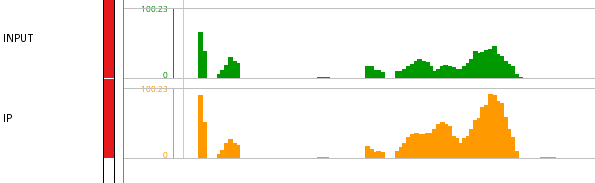
對于上述多種歸一化方式,其實都是可以拿來在樣本間比較的。在實際操作中,由于RPKM的概念最為經典,應用的也最為廣泛。
以上是“如何實現bigwig歸一化”這篇文章的所有內容,感謝各位的閱讀!相信大家都有了一定的了解,希望分享的內容對大家有所幫助,如果還想學習更多知識,歡迎關注億速云行業資訊頻道!
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





