溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這篇文章將為大家詳細講解有關DESeq2中怎么實現兩組間差異分析操作,文章內容質量較高,因此小編分享給大家做個參考,希望大家閱讀完這篇文章后對相關知識有一定的了解。
讀取基因的表達量表格和樣本的分組信息兩個文件,其中表達量的文件示例如下
gene_id ctrl-1 ctrl-2 ctrl-3 case-1 case-2 case-3 geneA 14 0 11 4 0 12 geneB 125 401 442 175 59 200
每一行為一個基因,每一列代表一個樣本。
分組信息的文件示例如下
sample condition ctrl-1 control ctrl-2 control ctrl-3 control case-1 case case-2 case case-3 case
第一列為樣本名,第二列為樣本的分組信息。
讀取文件的代碼如下
# 讀取表達量的表格 count <- read.table( "gene.counts.tsv", header=T, sep="\t", row.names=1, comment.char="", check.names=F) # 預處理,過濾低豐度的數據 countData <- count[apply(count, 1, sum) > 0 , ] # 讀取樣本分組信息 colData <- read.table( "sample.group.tsv", header=T, sep="\t", row.names=1, comment.char="", check.names=F) # 構建DESeq2中的對象 dds <- DESeqDataSetFromMatrix( countData = countData, colData = colData, design = ~ condition) # 指定哪一組作為control dds$condition <- relevel(dds$condition, ref = "control")
在讀取數據的過程中,有兩點需要注意,第一個就是根據表達量對基因進行過濾,通常是過濾低表達量的基因,這一步是可選的,閾值可以自己定義;另外一個就是指定哪一組作為control組,在計算log2FD時 ,需要明確control組,默認會字符串順序對分組的名字進行排序,排在前面的作為control組,這種默認行為選出的control可能與我們的實驗設計不同,所以必須明確指定control組。
計算每個樣本的歸一化系數,代碼如下
dds <- estimateSizeFactors(dds)
將原始的表達量除以每個樣本的歸一化系數,就得到了歸一化之后的表達量。
DESeq2假定基因的表達量符合負二項分布,有兩個關鍵參數,總體均值和離散程度α值, 如下圖所示
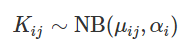
這個α值衡量的是均值和方差之間的關系,表達式如下
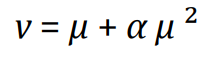
通過下列代碼估算每個基因的α值
dds <- estimateDispersions(dds)
代碼如下
dds <- nbinomWaldTest(dds) res <- results(dds)
為了簡化調用,將第二部到第四部封裝到了DESeq這個函數中,代碼如下
dds <- DESeq(dds) res <- results(dds) write.table( res, "DESeq2.diff.tsv", sep="\t", quote=F, col.names = NA)
在DESeq2差異分析的過程中,已經考慮到了樣本之間已有的差異,所以可以發現,最終結果里的log2FD值和我們拿歸一化之后的表達量計算出來的不同, 示意如下
> head(results(dds)[, 1:2]) log2 fold change (MLE): condition B vs A DataFrame with 6 rows and 2 columns baseMean log2FoldChange <numeric> <numeric> gene1 7.471250 -0.8961954 gene2 18.145279 0.4222174 gene3 2.329461 -2.3216915 gene4 165.634256 -0.1974001 gene5 38.300621 1.3573162 gene6 7.904819 1.8384322
提取歸一化之后的表達量,自己計算baseMean和logFoldChange, 示例數據包含了12個樣本,其中前6個樣本為control, 后6個樣本為case , 代碼如下
> nor_count <- counts(dds, nor = T)
> res <- data.frame(
baseMean = apply(nor_count, 1, mean),
log2FoldChange = apply(nor_count, 1, function(t){ mean(t[7:12]) / mean(t[1:6])})
)
> head(res)
baseMean log2FoldChange
gene1 7.471250 0.5380191
gene2 18.145279 1.3404422
gene3 2.329461 0.1991979
gene4 165.634256 0.8719078
gene5 38.300621 2.5621035
gene6 7.904819 3.5365201對比DESeq2和自己計算的結果,可以發現,baseMeans是一致的,而log2Foldchange 差異很大,甚至連數值的正負都發生了變化。
log2FD 反映的是不同分組間表達量的差異,這個差異由兩部分構成,一種是樣本間本身的差異,比如生物學重復樣本間基因的表達量就有一定程度的差異,另外一部分就是我們真正感興趣的,由于分組不同或者實驗條件不同造成的差異。
關于DESeq2中怎么實現兩組間差異分析操作就分享到這里了,希望以上內容可以對大家有一定的幫助,可以學到更多知識。如果覺得文章不錯,可以把它分享出去讓更多的人看到。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





