溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
本篇文章為大家展示了KEGGgraph怎樣根據kgml 文件從pathway中重構出基因互作網絡,內容簡明扼要并且容易理解,絕對能使你眼前一亮,通過這篇文章的詳細介紹希望你能有所收獲。
KEGGgraph 包可以解析kgml 文件,從中得到不同對象之間的網絡結構,并在此基礎上進一步挖掘其中的信息。
KEGGgraph 包提供了以下3種基本功能:
用法示例
# 讀取hsa00020xml 文件
> mapkG <- parseKGML2Graph("hsa00020.xml",expandGenes=TRUE, genesOnly = TRUE)
> mapkG
A graphNEL graph with directed edges
Number of Nodes = 30
Number of Edges = 101
> nodes(mapkG)
[1] "hsa:1738" "hsa:4967" "hsa:55753" "hsa:1743" "hsa:8801" "hsa:8802"
[7] "hsa:8803" "hsa:3417" "hsa:3418" "hsa:3419" "hsa:3420" "hsa:3421"
[13] "hsa:47" "hsa:2271" "hsa:48" "hsa:50" "hsa:1431" "hsa:4190"
[19] "hsa:4191" "hsa:5091" "hsa:5160" "hsa:5161" "hsa:5162" "hsa:1737"
[25] "hsa:5105" "hsa:5106" "hsa:6389" "hsa:6390" "hsa:6391" "hsa:6392"
> edges(mapkG)
$`hsa:1738`
[1] "hsa:4967" "hsa:55753" "hsa:5160" "hsa:5161" "hsa:5162" "hsa:1737"
$`hsa:4967`
[1] "hsa:3419" "hsa:3420" "hsa:3421" "hsa:3417" "hsa:3418"
$`hsa:55753`
[1] "hsa:3419" "hsa:3420" "hsa:3421" "hsa:3417" "hsa:3418"在 parseKGML2Graph 中,有兩個參數,expandGenes 和 genesOnly。
expandGenes 控制是否將基因進行展開,在pathway 中,會有1個KO 對應多個gene的情況,比如下面這種
<entry id="32" name="hsa:8801 hsa:8802 hsa:8803" type="gene" reaction="rn:R00405" link="">http://www.kegg.jp/dbget-bin/www_bget?hsa:8801+hsa:8802+hsa:8803">; <graphics name="SUCLG2, G-SCS, GBETA, GTPSCS..." fgcolor="#000000" bgcolor="#BFFFBF" type="rectangle" x="260" y="574" width="46" height="17"/> </entry>
expandGenes = TRUE 表示將基因展開,每個基因作為一個節點。
genesOnly 參數控制是否將其他類型的entry (比如compound等類型)展現在network 中,默認值為 TRUE,所以最終得到的network 中節點全部是基因。
通過parseKGML2Graph 這一步我們就可以從一張pathway 中得到基因產物(蛋白)的互作網絡, 還需要注意一點,整個網絡是一個有向圖, 因為基因產物之間的互作關系是由方向性的。
由于自帶的可視化不夠美觀,我們把nodes和edges 寫入文件,用cytoscape 進行可視化,用法示例
mapkNodes <- nodes(mapkG)
mapkEdges <- edges(mapkG)
mapkEdges <- mapkEdges[sapply(mapkEdges, length) > 0]
res <- lapply(1:length(mapkEdges), function(t){
name <- names(mapkEdges)[t]
len <- length(mapkEdges[[t]])
do.call(rbind, lapply(1:len, function(n){
c(name, mapkEdges[[t]][n])
}))
})
result <- data.frame(do.call(rbind, res))
write.table(result, "edges.txt", sep = "\t", row.names = F, col.names = F, quote = F)
write.table(mapkNodes, "nodes.txt", sep = "\t", row.names = F, col.names = F, quote = F)導入cytoscape 畫出來的圖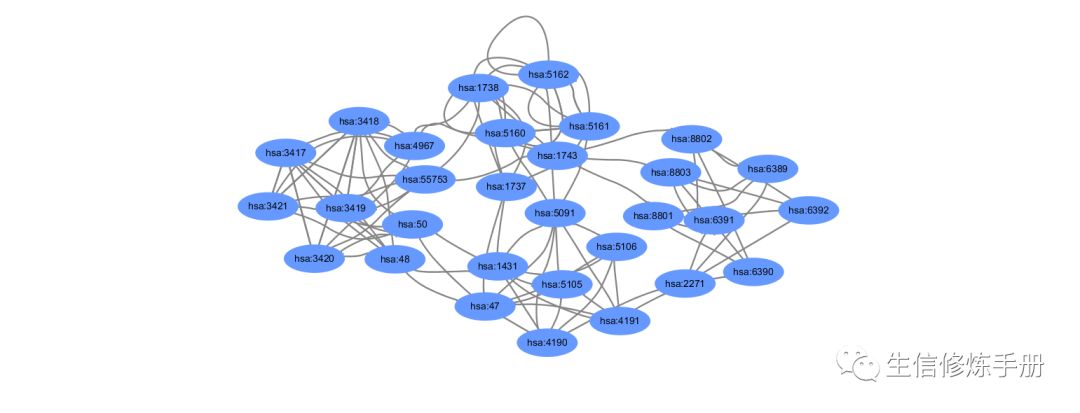
degree信息對于一個netwrok 而言,每個節點的degree 信息是我們最常用的信息, 示例
> mapkGoutdegrees <- sapply(edges(mapkG), length) > mapkGindegrees <- sapply(inEdges(mapkG), length) > degrees <- data.frame(indegrees = mapkGindegrees, outdegrees = mapkGoutdegrees) > head(degrees) indegrees outdegrees hsa:1738 1 6 hsa:4967 2 5 hsa:55753 2 5 hsa:1743 3 3 hsa:8801 4 1 hsa:8802 4 1
由于是有向圖,所以有入度 indegrees 和 出度 outdegrees 的概念。
除了以上基礎功能外,還可以借助其他的R包進一步挖掘信息,比如在整個基因互作網絡, 哪個基因是最關鍵的。
示例:
> library(RBGL)
> mapkG <- parseKGML2Graph("hsa00020.xml",expandGenes=TRUE, genesOnly = TRUE)
> bcc <- brandes.betweenness.centrality(mapkG)
> rbccs <- bcc$relative.betweenness.centrality.vertices[1L,]
> toprbccs <- sort(rbccs,decreasing=TRUE)[1:4]
> toprbccs
hsa:1743 hsa:2271 hsa:1738 hsa:47
0.21597893 0.16177167 0.14965648 0.09880362對于network 而言,我們一般認為degree 越大的點在這個網絡中越重要,所以需要看節點的degree 信息。除了這種基本的認識外,還有很多成熟的算法,從network 中挖掘關鍵節點。 RBGL 包提供了Brandes 的算法,用來衡量節點在網絡中的重要性,上面的結果中,toprbccs 就是我們篩選出的4個比較重要的節點。
使用KEGGgraph包,我們可以方便的從pathway中得到基因戶做網絡;
可以將network 中的nodes和edges 信息導出,使用cytoscape 可視化;
可以借助其他成熟的算法挖掘基因互作網絡中的關鍵基因;
上述內容就是KEGGgraph怎樣根據kgml 文件從pathway中重構出基因互作網絡,你們學到知識或技能了嗎?如果還想學到更多技能或者豐富自己的知識儲備,歡迎關注億速云行業資訊頻道。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





