溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
這期內容當中小編將會給大家帶來有關宏基因組binning的原理是什么,文章內容豐富且以專業的角度為大家分析和敘述,閱讀完這篇文章希望大家可以有所收獲。
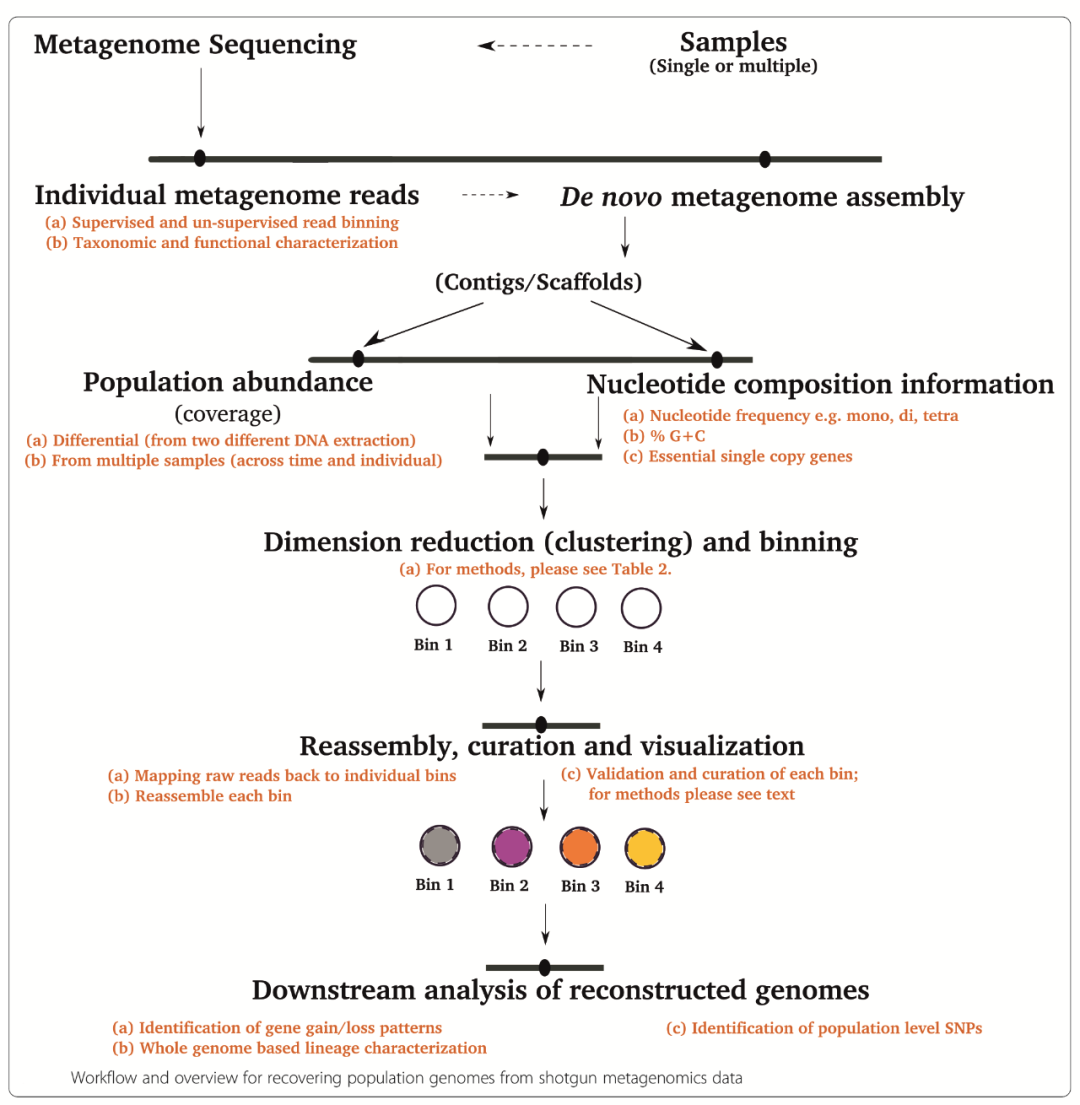
在宏基因組中分離單基因組,可利用序列特征或序列組裝信息,常見的可用信息主要有以下幾種:
a.根據核酸使用頻率(通常是四核苷酸頻率)、GC含量和必需的單拷貝基因等基因組特征;
b.根據contig序列的覆蓋度coverage信息;
c.根據測序數據的kmer豐度信息;
d.根據序列在不同樣品的共出現規律(co-abundance patternsacross multiple samples);
e.將序列map到數據庫的參考序列所獲得的注釋信息,也即物種binning。
根據所使用的序列數據不同,binning策略可分為三種:基于組裝前的clean reads,基于組裝后的contigs,基于注釋的基因genes。
⑴基于reads binning
環境樣本中微生物的豐度不同,其基因組kmer的期望深度也不同,根據kmer豐度可以直接對reads進行聚類,將屬于不同基因組的reads分離開來。其優勢是可以聚類出宏基因組中豐度非常低的物種,而且可以分離系統發育關系很近的物種。考慮到在宏基因組組裝中reads利用率很低,單樣品5Gb測序量情況下,環境樣品組裝reads利用率一般只有10%左右,腸道樣品或極端環境樣品組裝reads利用率一般能達到30%,這樣很多物種,尤其是低豐度的物種的reads沒有被沒有被組裝出來,沒有體現在contig中而被浪費,因此基于reads binning才有可能得到低豐度的物種基因組的的測序數據,在實際研究中基于reads binning的LSA(Latent Strain Analysis)方法可以聚類出豐度低到0.00001%的物種,并且對同一物種中的不同菌株的敏感性很強[2]。
⑵基于genes binning
在宏基因組做完序列組裝和基因預測之后,把所有樣品中預測到的基因混合在一起,去冗余得到unique genes集合,根據gene在各個樣品中的豐度變化模式,計算gene之間的相關性,利用這種相關性進行聚類。利用這種策略進行binning得到的bins可稱為CAG(co-abundance genegroups),包含有700個以上的gene的CAG稱為MGS(metagenomic species),CAG可用進行關聯分析,MGS可用進行后續的單菌組裝[3]。當然根據具體的聚類算法和相關性系數的不同,對genes binning得到的bins的叫法也不同,除以上外還有MLG(metagenomic linkage groups)、MGC(metagenomic clusters)和MetaOTUs(metagenomic operational taxonomicunits)等,同時,MLG, MGC, MGS和MetaOTUs物種注釋的標準也是不一樣的。
目前已發表的宏基因組關聯分析(MWAS)和多組學聯合分析文章中,宏基因組binning很多都用genes binning方法,尤其是疾病的MWAS研究中基本都用genes binning[4]。這種方法的優勢是基于genes豐度變化模式進行binning可操作性比較強,過程比較簡單,可復制性強,對計算機資源消耗比較低。
⑶基于contigs binning
在宏基因組做完序列組裝之后,將所有reads序列map到contigs上獲得contig覆蓋率,再綜合GC含量、核算組成等信息對contig進行聚類,將屬于不同基因組的contig序列分開。contig binning目前應用十分廣泛,最常用的就是用于組裝單物種基因組,目前已經有多種基于contig binning的軟件[1],對于豐度較高的物種contigs binning效果較好,但是目前也有些缺陷或者說還有很多可提升的空間,例如對核酸組成信息的利用,開發得就不夠充分,四堿基使用頻率因簡單而被廣泛使用和接受,但現在已有研究表明k-mer豐度信息也是很好的種系特征,同時越長的k-mer含有越多的信息,還有基因和參考基因組間的同源關系也是有價值的種系信號,但這些都還沒有被自動化的binning軟件整合。
上述就是小編為大家分享的宏基因組binning的原理是什么了,如果剛好有類似的疑惑,不妨參照上述分析進行理解。如果想知道更多相關知識,歡迎關注億速云行業資訊頻道。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





