溫馨提示×
您好,登錄后才能下訂單哦!
點擊 登錄注冊 即表示同意《億速云用戶服務條款》
您好,登錄后才能下訂單哦!
本篇文章給大家分享的是有關miRNA定量原理是什么,小編覺得挺實用的,因此分享給大家學習,希望大家閱讀完這篇文章后可以有所收獲,話不多說,跟著小編一起來看看吧。
在轉錄組的數據分析中,定量和差異分析是基礎分析內容,對于mRNA的定量,直接將reads比對參考基因組進行定量即可,但是對于miRNA數據而言,這樣的操作方式就不合適了。
miRNA長度在18bp-36b之間,非常的短,如果用這么短的reads比對整個基因組,肯定會有多個比對上的位置,在定量時,無法有效區分reads到底來自于哪一個miRNA,而且gtf文件中只會記錄miRNA基因的基因組位置,并不提供5’和3’端兩個成熟的miRNA的基因組位置。
為了準確對miRNA進行定量,mirdeep軟件的開發者提出了這樣一種思路,將reads 比對miRNA的前體序列,這樣可以有效判斷reads到底屬于哪一個miRNA;其次,考慮到一個miRNA前體可以產生兩個成熟的miRNA,根據reads的比對位置來區分兩個成熟的miRNA, 示意圖如下
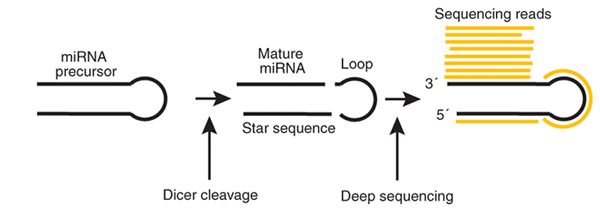
在最開始研究miRNA的時候,發現一個miRNA前體可以產生兩個小的RNA, 將其中表達量高的一個稱之為mature miRNA, 將表達量相對較低的稱之為star miRNA, 在后來直接將兩個RNA都稱之為mature miRNA了,mirdeep軟件中依然采用了star序列的命名方式,實際是都是miRNA的序列。
在實際操作時,通過bowtie1進行比對,bowtie1對于短序列的比對任務靈敏度更高,適用于miRNA這種短序列的比對工作,首先對miRNA前體構建索引,代碼如下
bowtie-build precursor.converted miRNA_precursor
接下來,將成熟的miRNA比對到miRNA前體上,代碼如下
bowtie -p $threads -f -v 0 -a --best --strata --norc miRNA_precursor mature.converted ${name1}_mapped.bwt最后,將miRNA測序的reads比對到miRNA前體上,代碼如下
bowtie -p $threads -f -v $mismatches -a --best --strata --norc miRNA_precursor $name2.converted ${name2}_mapped.bwt根據兩個比對產生的結果文件,就可以對miRNA進行定量,首先從成熟miRNA的比對結果中,確定成熟miRNA在miRNA前體上的位置, 比對結果示意如下
hsa-miR-550b-2-5p + hsa-mir-550b-1 15 ATGTGCCTGAGGGAGTAAGACA IIIIIIIIIIIIIIIIIIIIII 1 hsa-miR-550b-3p + hsa-mir-550b-1 57 TCTTACTCCCTCAGGCACTG IIIIIIIIIIIIIIIIIIII 1
對于hsa-mir-550b-1這個前體而言,有hsa-miR-550b-2-5p和hsa-miR-550b-3p兩個成熟的miRNA,hsa-miR-550b-2-5p比對到了前體的15-36bp的位置,hsa-miR-550b-3p比對到前體的57-76bp的位置,這個位置是根據第4列的值加上第五列的序列長度得到的。
接下來從測序reads的比對結果,確定每個miRNA的表達量,比對結果示意如下
SW1_224845_x3 + hsa-mir-550b-1 17 GTGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIIII 4 18:C>G SW1_287750_x2 + hsa-mir-550b-1 18 TGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIII 4 17:C>G SW2_152540_x1 + hsa-mir-550b-1 17 GTGCCTGAGGGAGTAAGACT IIIIIIIIIIIIIIIIIIII 2 19:A>T SW2_152541_x7 + hsa-mir-550b-1 17 GTGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIIII 4 18:C>G SW2_199319_x1 + hsa-mir-550b-1 18 TGCCTGAGGGAGTAAGAA IIIIIIIIIIIIIIIIII 4 17:C>A SW3_404761_x1 + hsa-mir-550b-1 17 GTGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIIII 4 18:C>G SW3_516112_x1 + hsa-mir-550b-1 18 TGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIII 4 17:C>G SW4_451132_x4 + hsa-mir-550b-1 17 GTGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIIII 4 18:C>G SW4_589021_x1 + hsa-mir-550b-1 18 TGCCTGAGGGAGTAAGAGA IIIIIIIIIIIIIIIIIII 4 17:C>G
我們可以看到,在實際測序的樣本中,有這么多reads比對到了hsa-mir-550b-1這個前體;接下來的任務就是區分這些reads到底屬于哪些成熟的miRNA。
從比對結果可以很明顯看到,實際測序的reads在前體上的位置都是在17-36bp之間,對于這個區間而言,只能是hsa-miR-550b-2-5p這個成熟的miRNA。
為了加快比對過程,mirdeep對測序reads進行了處理,合并了冗余reads, 用序列標識符中x后面的數字代表在原始測序結果中該reads出現的次數,最終計數時,直接把x后面對應的數字相加就可以了。
mirdeep軟件是miRNA分析中非常經典的一款軟件,了解其定量原理有助于我們更好的理解miRNA數據分析過程。
以上就是miRNA定量原理是什么,小編相信有部分知識點可能是我們日常工作會見到或用到的。希望你能通過這篇文章學到更多知識。更多詳情敬請關注億速云行業資訊頻道。
免責聲明:本站發布的內容(圖片、視頻和文字)以原創、轉載和分享為主,文章觀點不代表本網站立場,如果涉及侵權請聯系站長郵箱:is@yisu.com進行舉報,并提供相關證據,一經查實,將立刻刪除涉嫌侵權內容。





